Los secretos de la fibrosis pulmonar idiopática
La disfunción de las células epiteliales se postula como un componente importante en la patogénesis de la fibrosis pulmonar idiopática (FPI). Se han encontrado mutaciones en el gen de la proteína surfactante C (SP-C) (SFTPC), una proteína alveolar de tipo II (AT2), en la FPI esporádica y familiar. Para vincular causalmente estos eventos, recientes estudios han trabajado en un modelo animal capaz de regular la expresión de una sustitución de isoleucina a treonina asociada a la FPI en el codón 73 (I73T) del gen Sftpc (SP-CI73T). Las cohortes SP-CI73T tratadas con tamoxifeno desarrollan aumentos rápidos en el ARNm de SftpcI73T y en la proteína proSP-CI73T mal procesada, lo que se acompaña de mayor mortalidad temprana. Esta fase aguda se caracteriza por lesión pulmonar del parénquima difuso, infiltración tisular por monocitos, alveolitis policelular, y elevaciones en el lavado broncoalveolar y en el contenido de ARNm de citoquinas inflamatorias en células AT2. La resolución de la alveolitis acorde con un aumento en TGF-β1, es seguida por una remodelación aberrante marcada por deposición de colágeno, hiperplasia de células AT2 y fisiología pulmonar restrictiva. La relevancia traslacional del modelo se sustenta por la detección de múltiples biomarcadores de FPI previamente reportados en cohortes humanas. Estos datos demuestran que la expresión in vivo del SP-C mutante causa fibrosis pulmonar espontánea, lo que refuerza el papel de la disfunción de células AT2 como un factor clave de la patogénesis de la FPI.
Células AT2
El término diagnóstico fibrosis pulmonar idiopática (FPI) incluye la creencia de larga data de que esta enfermedad progresiva surge espontáneamente y que su causa es desconocida. Sin embargo, Nureki y sus colegas (DOI: 10.1172/JCI99287) han desafiado recientemente este concepto fundamental al crear un modelo translacional que puede acelerar el descubrimiento de fármacos para la FPI, patología que a menudo causa la muerte en un plazo de 3 a 5 años.
La FPI tiene un fenotipo complejo que se manifiesta por heterogeneidad clínica, etiológica y molecular. De hecho, se requiere heterogeneidad en las características radiográficas y patológicas de la neumonía intersticial habitual para el diagnóstico definitivo. Se han asociado con la FPI variantes raras y comunes en siete genes, incluyendo MUC5B, y al menos 12 loci, así como exposiciones ambientales (por ejemplo, asbesto y microorganismos) y condiciones autoinmunes (por ejemplo, artritis reumatoide y esclerodermia). Cada uno de estos factores asociados aumenta el riesgo de desarrollar las características radiográficas y patológicas de la neumonía intersticial habitual.
Los factores de riesgo más comunes de la FPI, como la edad avanzada, el sexo masculino, tabaquismo y la variante promotora MUC5B, también confieren una predisposición al desarrollo de una enfermedad similar a la FPI, incluida la enfermedad pulmonar intersticial asociada a la artritis reumatoide y la neumonitis por hipersensibilidad crónica. La heterogeneidad biológica de la FPI es aún más destacable por los múltiples fenotipos epigenéticos y transcripcionales moleculares emergentes de esta enfermedad. La heterogeneidad clínica, etiológica y molecular colectiva de la FPI sugiere que esta enfermedad representa una respuesta a las lesiones ambientales y endógenas recurrentes en un huésped susceptible, en el que la respuesta fibrótica progresiva no puede resolverse debido a defectos en uno o varios mecanismos clave implicados en la homeostasis pulmonar. En consecuencia, la integración de factores de riesgo clave (susceptibilidad genética, exposiciones ambientales y autoinmunidad) con mecanismos conocidos de la enfermedad, como la defensa del huésped, senescencia celular, función de las células broncoalveolares y la reparación pulmonar, podría establecer una guía de trabajo para un tratamiento más eficaz de la enfermedad precozmente establecida (figura 1).
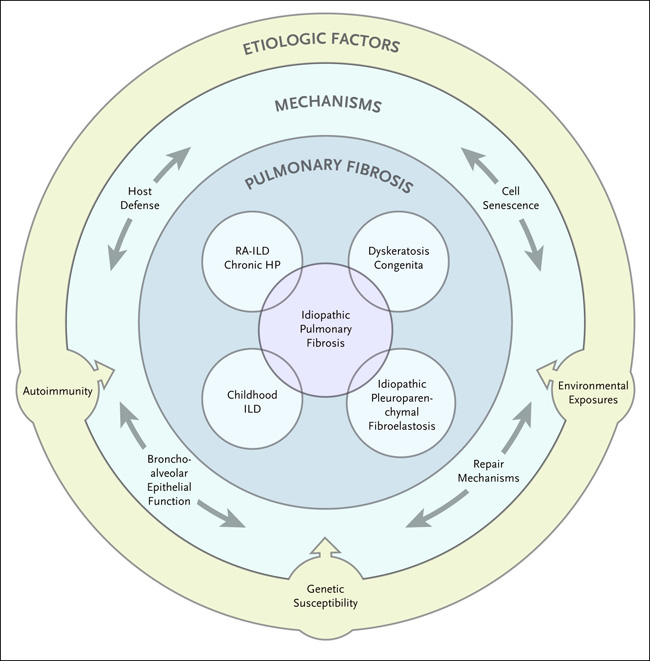
Figura 1. Hacia una comprensión de la fibrosis pulmonar idiopática (FPI), una enfermedad multifactorial.
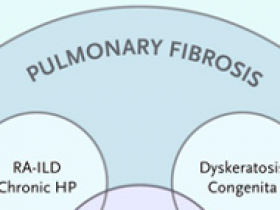
La FPI, una enfermedad heterogénea, es causada por anomalías en la defensa del huésped, la senescencia celular, función de las células broncoalveolares y reparación pulmonar, todas ellas afectadas por la autoinmunidad, las variantes genéticas y las exposiciones ambientales. Aunque cada uno de estos procesos biológicos puede fallar sustancialmente y resultar en tipos únicos de fibrosis pulmonar (enfermedad pulmonar intersticial reumatoide asociada a la artritis [RA-ILD], neumonitis por hipersensibilidad crónica [HP], enfermedad pulmonar intersticial infantil [ILD]), disqueratosis congénita, o fibroelastosis pleuroparenquimatosa idiopática), la mayoría de los casos de FPI probablemente ocurren en pacientes con defectos leves en estos mecanismos de homeostasis pulmonar, lo que resulta en una reparación ineficaz y una fibrosis pulmonar acumulativa que empeora con el tiempo.
Nureki y sus colegas se centraron en la fibrosis pulmonar asociada con una variante del gen de la proteína C del surfactante (SFTPC). Las mutaciones SFTPC son inusuales en pacientes con FPI y se observan con mayor frecuencia en niños con enfermedad pulmonar intersticial o en familias con antecedentes de inicio temprano de enfermedad pulmonar intersticial. Sin embargo, comprender cómo estas mutaciones causan la fibrosis pulmonar podría, en general, facilitar la comprensión de la patogénesis de la enfermedad. Los investigadores introdujeron una sustitución por missense (1286T→C) en secuencias ortólogas de ratón de SFTPC (SftpcI73T), regulando la expresión de este gen mutante en células epiteliales alveolares de tipo 2 (AT2), que secretan proteínas surfactantes. Esta rara variante de ganancia de función es la mutación SFTPC más común en humanos con enfermedad pulmonar intersticial asociada a SFTPC.
El estudio mostró que esta sustitución resultó en el desarrollo espontáneo de fibrosis pulmonar, presumiblemente causada por alteración del tráfico intracelular de la proteína C del surfactante, proteostasis defectuosa (mantenimiento de niveles de proteína a través del plegamiento, tráfico y degradación), alteración de la mitofagia y la mejora de la macroautofagia (pero no de la apoptosis) de las células AT2. Estos eventos moleculares se asociaron con el desarrollo espontáneo de alveolitis aguda con sobreexpresión de biomarcadores de FPI así como con remodelación fibrótica, incluyendo hiperplasia de células AT2, pero sin características específicas de la neumonía intersticial habitual. Estos hallazgos resaltan el papel potencial de las células de la proteína C del surfactante y células AT2 en el desarrollo de la fibrosis pulmonar, y al seguir estrategias genómicas funcionales, estos investigadores han desarrollado un modelo espontáneo de enfermedad pulmonar intersticial que puede resultar útil como plataforma traslacional para la FPI.
La proteína C del surfactante es un péptido hidrófobo con funciones relacionadas con su capacidad, en presencia de fosfolípidos adecuados, para reducir la tensión superficial a bajos volúmenes pulmonares facilitando la difusión y adsorción de los fosfolípidos existentes en la interfaz aire-líquido. La proteína se inserta en la monocapa de fosfolípidos en función de la tensión superficial, facilitando la diseminación después de la compresión alveolar a bajos volúmenes, promoviendo el reciclaje del surfactante. Muchos investigadores han sugerido que el colapso alveolar y la induración destán implicados en la patogénesis de la fibrosis pulmonar, y aumentan en ausencia de un surfactante que funcione normalmente o de una producción deficiente de células AT2 o del reciclado del surfactante. El estrés crónico fisiológico y broncoalveolar por apertura y cierre cíclico de alvéolos atelectásicos o por sobredistención de alvéolos adyacentes a áreas de induración es más pronunciado en los lóbulos periféricos y lóbulos inferiores del pulmón y, en consecuencia, puede explicar la distribución de las lesiones fibróticas en la FPI.
El intrigante trabajo de Nureki y sus colegas ha solidificado algunos de los conceptos básicos de la fibrosis pulmonar y ha creado un modelo traslacional de la enfermedad pulmonar fibrótica; este trabajo destaca la importancia de los blancos genéticos en la comprensión de las causas y la patogénesis de esta enfermedad. Cabe destacar que su investigación ha reforzado la importancia de la lesión de las células AT2 en las etapas iniciales de la fibrosis pulmonar. El trabajo también muestra que los defectos endógenos en la función celular, así como los defectos inducidos por exposiciones ambientales, pueden causar lesiones microscópicas recurrentes en el espacio alveolar, y que las células AT2 que funcionan mal pueden servir para iniciar y perpetuar el proceso fibroproliferativo. Aunque ningún modelo murino ha recapitulado completamente la heterogeneidad compleja y la patogénesis de la FPI, el modelo espontáneo de la enfermedad pulmonar intersticial asociada al SFTPC creado por Nureki y sus colegas probablemente resulte valioso para comprender la biología de la fibrosis pulmonar y desarrollar nuevos fármacos para esta enfermedad progresiva.
Fuente bibliográfica
Revealing the Secrets of Idiopathic Pulmonary Fibrosis
Richard K. Albert, M.D., and David A. Schwartz, M.D.
Departments of Medicine (R.K.A., D.A.S.) and Microbiology and Immunology (D.A.S.), University of Colorado School of Medicine, Aurora.
DOI: 10.1056/NEJMcibr1811639
Temas Relacionados