Acoplamiento molecular para descubrir antivirales
En promedio, un medicamento aprobado cuesta actualmente entre 2.000 y 3.000 millones de dólares y tarda más de 10 años en desarrollarse. Esto se debe en parte a los costosos y largos experimentos en laboratorios, a los escasos compuestos que tienen éxito en pruebas iniciales y a las altas tasas de desgaste en las fases pre y clínicas. El cribado virtual basado en estructuras tiene el potencial de mitigar estos problemas. Mediante el screening estructural, la calidad de aciertos para encontrar moléculas de interés terapéutico mejora con el número de compuestos examinados. De hecho, se han llevado a cabo investigaciones utilizando modelos computacionales de acoplamiento molecular de la espícula (proteína S) del SARS-CoV-2, la cual interactúa con el receptor ya conocido, la enzima convertidora de angiotensina 2 (ECA2). De esta forma, se pueden identificar moléculas (potenciales antivirales) con afinidad por su ligando previamente estimada con la capacidad de interferir en la unión entre la proteína viral y ECA2. Esta plataforma tecnológica basada en el análisis de las estructuras moleculares y que ya ha sido utilizada para el descubrimiento de medicamentos como el nelfinavir contra el VIH, tiene el potencial de reducir considerablemente el tiempo que lleva el proceso de desarrollo de nuevos fármacos contra patógenos que no pierden tiempo en convertirse en una amenaza para la salud pública.
Ensemble docking
Actualmente, se necesitan urgentemente medicamentos eficaces para la enfermedad causada por el nuevo coronavirus (COVID-19), pero ¿cuál es la forma más rápida de encontrarlos? Un enfoque esperable es esperar que los medicamentos que han funcionado contra un virus diferente (como la hepatitis C o el Ébola) también funcionen contra el COVID-19. Alternativamente, se puede ser racional y enfocarse específicamente a las proteínas del SARS-CoV-2 para interrumpir su ciclo replicativo.
El genoma del SARS-CoV-2 codifica aproximadamente 25 proteínas que el virus necesita para infectar y para replicarse (figura 1). Entre ellas se encuentran la conocida proteína de la espícula (S), que reconoce la enzima convertidora de angiotensina 2 (ECA2) en la etapa inicial de la infección; dos proteasas, que cortan las proteínas víricas y humanas; la ARN polimerasa, que sintetiza el ARN viral; y una endorribonucleasa que corta el ARN. Encontrar medicamentos que puedan unirse a las proteínas virales y evitar que funcionen es una forma lógica de avanzar y la prioridad de muchos laboratorios de investigación.

Figura 1. El virus SARS-CoV-2 y sus proteínas.
Aunque todas las proteínas del SARS-CoV-2 son blancos farmacológicos potenciales, para algunas de ellas es más probable encontrar algún medicamento que sea eficaz, particularmente para aquellas que desempeñan las funciones principales en el ciclo replicativo del virus y para las que carecen de homólogos de proteínas humanas. Entre los ejemplos figuran la glucoproteína de espícula, las proteasas de tipo papaína y tipo quimotripsina y la ARN polimerasa dependiente de ARN. ACE2 denota la enzima convertidora de angiotensina 2; NSP, la proteína no estructural; ORF, marco de lectura abierto; y la RdRP, ARN polimerasa dependiente de ARN.
Un enfoque que puede ser de utilidad para encontrar fármacos dirigidos a componentes virales es el análisis de estructuras a nivel computacional (figura 2). En este proceso, computadoras "acoplan" los compuestos de prueba en sitios de unión en modelos tridimensionales de las proteínas objetivo. Las afinidades de unión de los compuestos se calculan con el uso de ecuaciones basadas en la física que cuantifican las interacciones entre el fármaco y su objetivo. A continuación, los compuestos mejor clasificados se someten a pruebas experimentales para comprobar si realmente se unen y tienen los efectos descendentes necesarios (como la detención de la infectividad viral) en células y modelos animales.
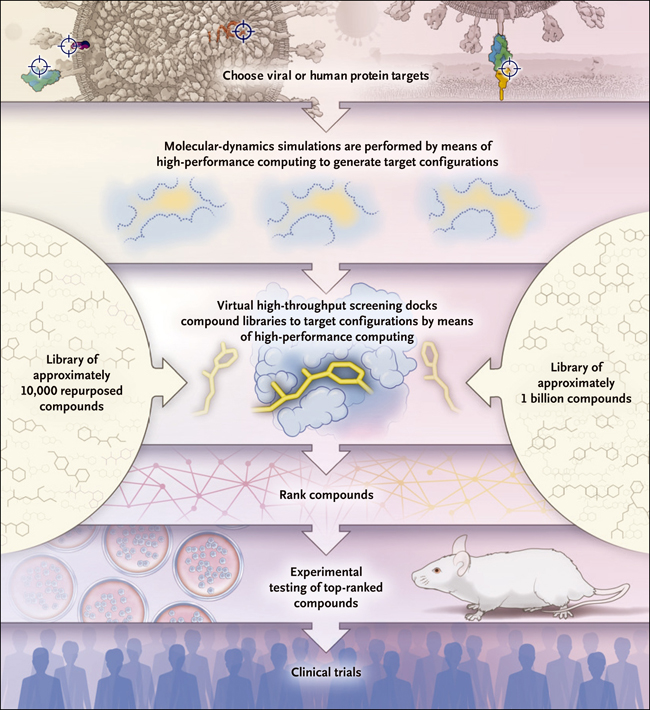
Figura 2. Acoplamiento molecular por ensamblaje de alto rendimiento vía computacional para el descubrimiento de fármacos.
Acoplar una biblioteca con fines específicos puede llevar al descubrimiento de compuestos de rápido despliegue. Las bibliotecas más grandes, que pueden contener miles de millones de compuestos, son útiles para descubrir rápidamente nuevos compuestos aún no probados en seres humanos.
El descubrimiento de medicamentos basados en estructuras ha sido importante para encontrar antivirales, un ejemplo de ello es el nelfinavir, descubierto en la década de 1990, para tratar la infección por el virus de la inmunodeficiencia humana (VIH). Sin embargo, lamentablemente, en ese momento el proceso era relativamente ineficiente: los cálculos eran inexactos y las computadoras eran tan débiles que sólo se podían acoplar unos 100 compuestos a la vez. Además, tanto el objetivo como el fármaco tenían que mantenerse rígidos en el proceso de acoplamiento en un enfoque de bloqueo. El acoplamiento rígido no suele tener lugar en la vida real, porque las proteínas se someten a movimientos internos impulsados por la temperatura que da lugar a formas fluctuantes de los sitios de unión.
Desde los años 90, la potencia de las supercomputadoras se ha multiplicado por un millón más o menos. Actualmente, se puede realizar acoplamiento rígido de más de mil millones de compuestos en unos pocos días. Así, el cribado virtual de alto rendimiento está superando al cribado experimental equivalente de alto rendimiento y puede identificar rápidamente compuestos muy estrechamente vinculados (Nature. 2020 Apr;580(7805):663-668). Además, se pueden realizar simulaciones de dinámica molecular para calcular los movimientos internos de las proteínas y se pueden examinar los medicamentos candidatos mediante un proceso que utiliza las diferentes formas del sitio de unión en un procedimiento conocido como "ensemble docking" que corresponde a la generación de un "ensamblaje" de conformaciones de blancos farmacológicos para el descubrimiento de medicamentos basándose en estructuras computacionales (Biophys J. 2018 May 22;114(10):2271-2278). Este enfoque es más realista que el acoplamiento rígido y ha tenido éxito, por ejemplo, en el descubrimiento de medicamentos contra el VIH a partir del 2000. Además, este tipo de acoplamiento molecular ha producido resultados validados experimentalmente contra diversos blancos de proteínas.
Las supercomputadoras modernas, como la Summit del Oak Ridge National Laboratory, que es actualmente la más potente del mundo, realizan un procesamiento masivo en paralelo en el que se llevan a cabo muchos cálculos simultáneamente. Esto permite que las simulaciones de la dinámica molecular de muchas réplicas del blanco se ejecuten en paralelo, cada una explorando un espacio conformacional ligeramente diferente. Así, con el uso de Summit en un día se puede obtener un modelo de simulación completo de un blanco farmacológico, en este caso para las proteínas del SARS-CoV-2, mientras que con el uso de un típico conglomerado de computadoras se tardaría meses. Las supercomputadoras también se utilizan en el acoplamiento rápido en paralelo de grandes bases de datos de compuestos. De esta manera, el campo de descubrimiento de medicamentos basado en estructuras está preparado para obtener resultados rápidos.
Entonces, ¿qué ocurre actualmente? La laboriosa y clásica vía de descubrimiento y aprobación de nuevos fármacos, que ha durado décadas, no podría ser menos adecuada para la actual pandemia. La reutilización de los medicamentos existentes ofrece un mecanismo potencialmente rápido de despliegue, ya que los perfiles de seguridad son conocidos. Por lo tanto, a mediados de febrero se publicó en un servidor de preimpresión un informe preliminar de un estudio de "ensemble docking" hecho por supercomputadoras sobre una base de datos de compuestos de contra la proteína S del virus, con 8.000 compuestos clasificados según la afinidad de unión calculada con el dominio de unión al receptor de la proteína S (ChemRxiv. Preprint. https://doi.org/10.26434/chemrxiv.11871402.v4). Los resultados informarán los futuros cálculos en un proceso rápido e iterativo.
Sin embargo, en el mundo surrealista y acelerado de la investigación contra COVID-19, los avances se desactualizan rápidamente. Se están notificando en rápida sucesión muchas nuevas estructuras tridimensionales experimentales de la proteína S y otros blancos virales, un proceso que requiere que las simulaciones y el acoplamiento se perfeccionen y se repitan. La inteligencia artificial se está utilizando para predecir la unión de los potenciales nuevos fármacos. En todo el mundo se han establecido diferentes tipos de programas experimentales de detección en laboratorios y están aumentando. Mientras tanto, para varias proteínas del SARS-CoV-2, la selección virtual de alto rendimiento y el acoplamiento se encuentra en modo de producción total, tanto en superordenadores como con el uso de vastos recursos de computación en nube. Nada de esto garantiza el éxito en un plazo determinado, pero una combinación de racionalidad, conocimiento científico e ingenio con las herramientas más poderosas disponibles nos dará nuestra mejor oportunidad.
Fuente bibliográfica
How to Discover Antiviral Drugs Quickly
Jerry M. Parks, Ph.D., and Jeremy C. Smith, Ph.D.
Department of Biochemistry and Cellular and Molecular Biology, University of Tennessee, Knoxville (J.C.S.).
DOI: 10.1056/NEJMcibr2007042
Temas Relacionados