Cardiología
Los monocitos en la aterosclerosis
En este nuevo estudio, los investigadores demuestran por qué los macrófagos permanecen en las placas arteriales y conducen finalmente a la aterosclerosis. Se sabe que las placas de las arterias que se desprenden tienen un alto contenido de macrófagos, pudiendo bloquear las arterias, generar infartos al corazón y derrames cerebrales,. La aterosclerosis es estimulada por estos macrófagos cargados de colesterol en la pared arterial. Normalmente, el sistema inmunitario envía macrófagos para limpiar los depósitos de colesterol en las arterias, pero una vez que están llenos con la forma no saludable de colesterol pueden quedarse atascados en las arterias, activando la respuesta inflamatoria del cuerpo.
Por otra parte, la proteína netrina-1 promueve la aterosclerosis mediante la retención de los macrófagos en la pared arterial. De hecho, el control de ciertas señales detienen la migración y, como resultado de esto, las células se acumulan dentro de la placa. Además, los experimentos señalan, que al eliminar genéticamente la netrina-1, se puede minimizar la aterosclerosis, reducir el nivel de macrófagos en la placa y promover su migración.
Monocitos y aterosclerosis
Durante generaciones la presión evolutiva ha perfeccionado el sistema inmunológico humano para combatir eficientemente los retos infecciosos. Sin embargo, el mismo sistema se puede volver en contra, por ejemplo, cuando se activa por ciertos estímulos nocivos, como en el caso de los alimentos ricos en colesterol desencadenantes de la aterosclerosis.
Los monocitos son generados en las paredes arteriales inflamadas por la acumulación subendotelial de lipoproteínas que contienen apolipoproteína B, donde se transforman en macrófagos. Éstos, toman el colesterol para dar lugar a células espumosas, las que impulsan la progresión de lesiones ateroscleróticas. Como los macrófagos ingieren y procesan lípidos, pueden contribuir al desarrollo de placas vulnerables a través de la formación de un núcleo necrótico y el adelgazamiento de la capa fibrosa. La ruptura de placa es responsable de la mayoría de los infartos agudos de miocardio de origen aterotrombótico, y los macrófagos se encuentran principalmente en los sitios de ruptura de la placa en las arterias coronarias trombosadas de los pacientes con infarto agudo de miocardio. Un reciente estudio de Janine M. van Gils (Nat Immunol 2012; 13:136-43) y colegas subraya el rol aterogénico de la acumulación de macrófagos en la pared de los vasos y el potencial terapéutico cuando se pueda prevenir.
Las primeras investigaciones sobre los mecanismos moleculares de la aterosclerosis se centraron en las señales responsables del reclutamiento de monocitos en las paredes de las arterias inflamadas. Esta migración se produce en respuesta a las quimiocinas - señales moleculares direccionales elaboradas por las células endoteliales que recubren los lípidos retenidos en la aterosclerosis temprana. La quimiocina CCL2 contribuye a este proceso, interactuando con su receptor, CCR2, que se expresa en los monocitos circulantes (fig. 1). Los ratones deficientes para CCL2 o sus receptores son resistentes al desarrollo de la aterosclerosis, y esta interacción ha sido objeto de considerable atención como potencial blanco para nuevos tratamientos farmacológicos.
Aunque los primeros trabajos se centraron en las señales que guiaban los leucocitos hacia los tejidos, es cada vez más evidente que su salida es también un proceso altamente regulado, lo que podría ser susceptible de manipulación terapéutica. Por ejemplo, la salida de los linfocitos de los tejidos linfoides es inducida por un quimioatrayente de lípidos llamado esfingosina-1-fosfato. La inhibición de esta vía por el principio activo fingolimod resulta en linfopenia periférica debido a la retención de los linfocitos en los ganglios linfáticos. Se cree que este mecanismo contribuye a la eficacia del fingolimod en pacientes con esclerosis múltiple, para lo que ha sido aprobado. Las investigaciones sugieren que la salida de los macrófagos de la pared de los vasos inflamados también está dirigida por distintos factores quimiotácticos que estimulan la entrada de monocitos, y que el fracaso de este proceso provoca la hipercolesterolemia, contribuyendo a la inflamación crónica y progresión de la aterosclerosis. El trasplante de arcos aórticos ateroscleróticos de animales con hipercolesterolemia a animales con niveles normales de colesterol permite la regresión de la placa en tejidos vasculares, y los estudios de seguimiento han demostrado que los macrófagos escapan de las lesiones en los vasos transferidos después de su exposición a niveles normales de colesterol. Análisis posteriores han demostrado que la salida de los macrófagos de las lesiones ateroscleróticas está dirigida por dos quimiocinas, CCL19 y CCL21, que interaccionan con su receptor, CCR7, en los macrófagos. En modelos animales, las drogas que activan el receptor nuclear LXR promueven la salida de monocitos de los vasos inflamados, al menos en parte, por la sobre-regulación de la expresión de macrófagos CCR7.
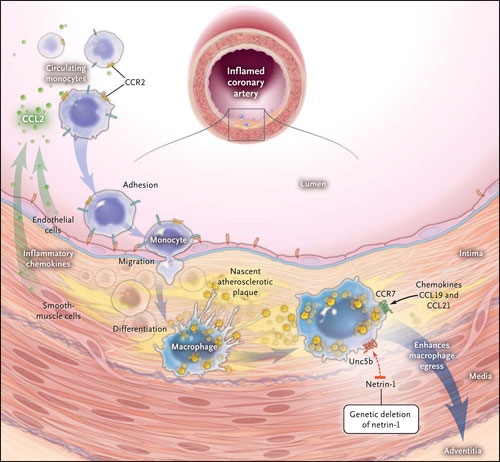
La adhesión de los monocitos y la acumulación en placa aterosclerótica naciente son provocadas por quimiocinas inflamatorias (como CCL2) activando a CCR2 en los monocitos circulantes. Los monocitos se diferencian dentro de los macrófagos, uniéndose al colesterol y transformándose en células espumosas. Pueden ser liberados de la placa aterosclerótica, probablemente en la adventicia circundante. Esta salida es estimulada por la acción de las quimiocinas CCL19 y CCL21 en CCR7. van Gils y colegas han identificado a la netrina-1 (que activa el receptor Unc5b) como un inhibidor de la salida de macrófagos. Se concluyó que la supresión genética de esta señal de parada aumenta el escape de los macrófagos del ateroma, limitando la progresión de la enfermedad.
El estudio realizado por el grupo de Janine M. van Gils se suma al conocimiento que los monocitos en lesiones ateroscleróticas bloquean la salida de los macrófagos de la pared de los vasos inflamados en la aterosclerosis. Ellos obtuvieron indicios del sistema nervioso en desarrollo, en el que ciertas neuronas pueden ser atraídas y repelidas posteriormente durante la compleja orquestación de movimiento celular necesaria para la ruta axonal. Una molécula particular de orientación neuronal, netrina-1, curiosamente sirve como un tope y señal de movimiento, dependiendo de la expresión de sus receptores en la superficie de las células diana. Sorprendentemente se ha observado que la netrina-1 es altamente expresada por macrófagos activados. Basándose en esta indicación, los autores encontraron que la netrina-1 inhibe la migración de monocitos, que de otro modo sería guiada por quimiocinas asociadas a la aterosclerosis, como CCL2. Esto llevó a la hipótesis que la netrina-1 es la señal de parada que bloquea la salida de los macrófagos de las paredes de los vasos inflamados en presencia de hipercolesterolemia. Este fracaso de los mecanismos normales de salida daría lugar a la inflamación crónica de la pared de los vasos y progresión de la placa.
En apoyo de este postulado, los investigadores encontraron que netrina-1 y uno de sus receptores, Unc5b, se expresan con firmeza en las placas ateroscleróticas y que tal expresión se incrementa con la carga de colesterol en humanos y en macrófagos de ratón. Por otra parte, la inhibición de netrina-1 en macrófagos induce la emigración de las lesiones ateroscleróticas, a pesar de la hipercolesterolemia en curso, disminuyendo fuertemente la aterosclerosis. Los mecanismos por los cuales la netrina-1 aturde a los monocitos lesionados siguen siendo poco conocidos. El grupo de van Gils obtuvo pistas adicionales que sugieren que la netrina-1 interfiere con una vía de señalización que implica la proteína Rac1, un modulador clave del citoesqueleto de actina, disposición que gobierna la motilidad celular.
Así, esta investigación ha identificado un mecanismo para el bloqueo de la salida de los macrófagos producida por hipercolesterolemia y por lo tanto, se ha identificado un potencial objetivo terapéutico para la aterosclerosis. Las estrategias que quieran explotar esta observación clínica probablemente se compliquen, ya que las señales deben ser entregadas con precisión para mover las células donde se quiere que vayan. Para manipular terapéuticamente la migración celular, se necesita diseñar la ubicación precisa de las señales, así como la magnitud de su expresión. Este aspecto es resaltado por resultados de otro estudio en el que se determinó que la administración intravenosa de netrina-1 a ratones con hipercolesterolemia resultó en una menor aterosclerosis, probablemente por el aumento de expresión de netrina-1 en células endoteliales y, por ende, se atraparon los monocitos antes de llegar a la placa en crecimiento. Por lo tanto, la netrina-1 puede tener una influencia diferencial sobre la inflamación, dependiendo del sitio de su expresión.
Dicho esto, el trabajo de van Gils y compañeros pone de relieve las oportunidades terapéuticas que pueden derivarse de una mejor comprensión de los mecanismos de salida de los monocitos. Las intervenciones que estimulen directamente la salida de los macrófagos de las placas serán capaces de crear una sinergia con las terapias hipolipemiantes para hacer más eficaz y rápida la regresión de la aterosclerosis.
Fuente bibliográfica
The Monocyte in Atherosclerosis — Should I Stay or Should I Go Now?
Robert E. Gerszten, M.D., and Andrew M. Tager, M.D.
Massachusetts General Hospital and Harvard Medical School
N Engl J Med 2012; 366:1734-1736