Controlando la progresión tumoral
La angiogénesis tumoral es la proliferación de una red de vasos sanguíneos capaces de penetrar en los tejidos cancerosos, suministrando nutrientes y oxígeno y eliminando productos de desecho. El proceso comienza cuando las células del cáncer liberan moléculas que envían señales a los receptores ubicados en los tejidos normales del entorno. Esta señalización activa ciertos genes en el tejido de acogida que, a su vez, producen proteínas para estimular el crecimiento de nuevos vasos sanguíneos.
El crecimiento de estos vasos en los tumores es sólo parte de la historia. En 1971 el doctor Judah Folkman postuló que la prevención de la neovascularización podría inhibir el crecimiento a través del control de nutrientes vitales. La existencia de inhibidores naturales de la angiogénesis se insinuó en una intrigante observación hecha por cirujanos. Ellos encontraron que la extirpación quirúrgica de un gran tumor primario suele conducir a la rápida evolución de crecimientos metastásicos. Esto sugiere que el tumor primario estaba produciendo algo que mantenía un reducido número de metástasis en progreso. Cuando la neoplasia era removida, los tumores más pequeños crecían libremente.
Los inhibidores de origen natural son pequeñas proteínas que se derivan de otras proteínas más grandes que, sorprendentemente, tienen diferentes funciones en el cuerpo. Por ejemplo, ensayos clínicos con endostatina para el tratamiento del cáncer se encuentran actualmente en curso. Los tratamientos anti-angiogénesis comparten dos características muy interesantes: 1) debido a que son productos naturales del cuerpo, deberían ser mucho menos tóxicos que la quimioterapia convencional. 2) como actúan en las células normales (vasos sanguíneos) en lugar de atacar directamente al tumor, es menos probable que conduzca a la selección de resistencia.
La formación de vasos sanguíneos, o la falta de ella, está en la base de muchas enfermedades humanas, el control de este proceso tiene un fuerte potencial en varios trastornos, además del cáncer.
Nuevas herramientas contra los vasos sanguíneos del tumor
La aprobación por la FDA (siglas para Food and Drug Administration) de bevacizumab, un anticuerpo monoclonal humanizado, contra el factor de crecimiento endotelial vascular (VEGF) ha generado un enorme revuelo entre los oncólogos. El bevacizumab, primer tratamiento contra el cáncer que actúa mediante el bloqueo de la angiogénesis, es el resultado de la exitosa traducción del trabajo de Judah Folkman y colegas, quienes propusieron que la vascularización es esencial para el crecimiento de carcinomas invasivos clínicamente relevantes. Otro estudio reciente realizado por Christian Fischer y colegas (Cell 2007; 131:463-75) indica que un anticuerpo monoclonal contra una proteína angiogénica, el factor de crecimiento placentario PlGF, también puede ser una terapia antiangiogénica útil para una neoplasia en los seres humanos.
La células tumorales producen y secretan, en diversos grados, muchos y diferentes factores angiogénicos, incluyendo VEGF, PlGF, células estromales derivadas del factor 1 y angiopoyetina 2. La transcripción de los genes que codifican estas proteínas está mediada a través del factor inducible por hipoxia 1, que se expresa en niveles elevados en muchos tumores como respuesta a la menor disponibilidad de oxígeno y como consecuencia de alteraciones genéticas que activan oncogenes y de la inactivación de los genes supresores de tumores.
Una vez producidos, los factores angiogénicos estimulan la angiogénesis por medio de dos mecanismos. En primer lugar, tienen efectos locales por su unión a receptores afines en las células endoteliales vasculares peritumorales. El receptor estimula estas células para participar en la gemación de nuevas ramas de capilares que suministran sangre a los vasos sanguíneos del tumor. En segundo lugar, los factores angiogénicos secretados se unen a receptores de células situadas en lugares distantes, incluyendo la médula ósea, y estimulan la movilización de éstas en la circulación y su posterior alojamiento en el tumor. Estas células promueven la vascularización, ya sea por su incorporación en los vasos sanguíneos incipientes o por la producción de factores angiogénicos adicionales.
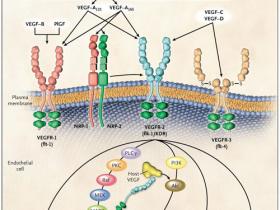
A pesar que se ha demostrado que PlGF y VEGF contribuyen a estos dos mecanismos, la evidencia sugiere que la unión de VEGF al receptor 2 de VEGF (VEGFR-2) es fundamental, ya que activa la proliferación de células endoteliales y la supervivencia. También es importante el enlace de PlGF al receptor 1 de VEGF (VEGFR-1) para la captación de macrófagos y otras células proangiogénicas derivadas de la médula ósea. La distinción funcional entre estas proteínas realza la observación de que VEGF es esencial para la normal vascularización embrionaria y el desarrollo, mientras que PlGF es prescindible para estos procesos.
Fischer y colegas informan que en los modelos de ratón para el cáncer, el tratamiento con anticuerpos anti-PlGF inhibe el crecimiento tumoral, la angiogénesis y la recaptación de macrófagos (que expresan VEGFR-1). En contraste, el tratamiento con anticuerpos anti-VEGFR-2 disminuye el crecimiento del tumor y la angiogénesis, pero no la integración de macrófagos. El reclutamiento de células mieloides es un importante mecanismo de resistencia tumoral a la terapia anti-VEGF. Como era de esperar, algunos modelos de tumores mostraron una mayor respuesta a la lucha contra anticuerpos VEGFR-2, mientras que otros respondieron mejor a la lucha contra el tratamiento PlGF. La terapia de combinación con anticuerpos da lugar a una mayor inhibición del crecimiento tumoral que la inhibición con anticuerpos solos, lo que sugiere que los dos anticuerpos tienen diferentes mecanismos de acción. El manejo de combinación también da lugar a una mayor inhibición de la linfangiogénesis que la inhibición con el tratamiento inicial solo. De este modo, tanto la metástasis vascular y como linfática pueden verse afectadas. Estos resultados proporcionan importantísimos datos preclínicos que pudiesen apoyar futuros ensayos con anticuerpos anti-PlGF humanizados.
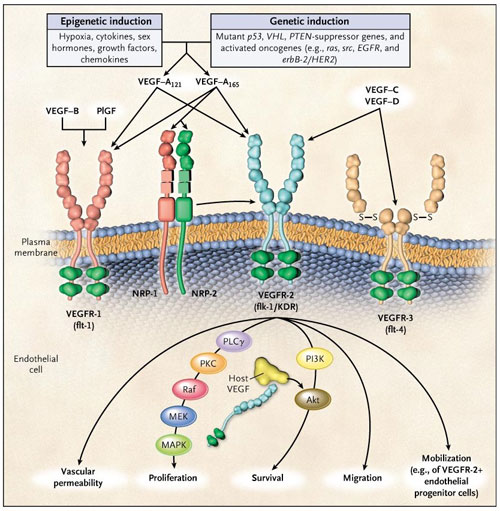
El principal mediador de la angiogénesis tumoral es el factor de crecimiento endotelial vascular A (VEGF-A, también llamado VEGF), específicamente sus isoformas circulantes VEGF121 y VEGF165. El papel de VEGFR-1 en la angiogénesis está mucho menos definido. VEGF se expresa en la mayoría de los cánceres humanos, y el aumento de expresión en los tumores a menudo se asocia con un pronóstico poco favorable. La inducción tumoral de VEGF puede ser causada por numerosas factores ambientales (epigenéticos) tales como la hipoxia, bajo pH, citoquinas inflamatorias (por ejemplo, interleucina-6), factores de crecimiento (como el factor de crecimiento de fibroblastos), hormonas sexuales (andrógenos y estrógenos) y quimiocinas (por ejemplo, células estromales derivadas de factor 1). Otras causas incluyen cambios genéticos inducidos tales como la activación de numerosos oncogenes o la pérdida o inactivación mutacional de una variedad de genes supresores de tumores. La unión de VEGF a VEGFR-2 conduce a una cascada de diferentes vías de señalización, generando la sobre regulación génica implicada en la mediación de la proliferación y migración de células endoteliales y en la mayor supervivencia y permeabilidad vascular (fotografía Robert S. Kerbel, N Engl J Med. 2008 May 8; 358(19):2039-49).
Sin embargo, los resultados observados en los estudios preclínicos con bevacizumab, han puesto de manifiesto los modestos efectos en posteriores ensayos clínicos, así como con otros agentes antiangiogénicos que no han demostrado eficacia en seres humanos. Las limitaciones se asocian con los modelos tumorales utilizados en los estudios preclínicos en los que un millón de células cancerosas se inyectan en el tejido subcutáneo (en un xenotransplante) u órgano de origen (por medio de trasplante ortotópico). El rápido crecimiento del tumor resultante induce a una enorme exigencia de angiogénesis. Este requisito no puede ser satisfecho por la activación de las células vasculares locales, ya que se requiere de la captación de células angiogénicas derivadas de la médula ósea. En contraste, los tumores que surgen espontáneamente en ratones con una mutación en el gen supresor PTEN o tratados con químicos cancerígenos se desarrollan más lentamente durante un largo período de tiempo y no dependen de la presencia de células procedentes de la médula ósea para la angiogenesis. La mayoría de los casos de cáncer en las personas se desarrollan lentamente en el transcurso de muchos años, por lo tanto parece probable que los modelos animales sobreestimen el efecto terapéutico de los agentes antiangiogénicos, especialmente los que bloquean el reclutamiento de células provenientes de la médula ósea.
En los ratones tratados con anticuerpos anti-PlGF, Fischer y colegas no observaron los efectos secundarios que suelen darse después del tratamiento con anticuerpos anti-VEGFR-2. Esto es consistente con la hipótesis de que, en comparación con VEGF, PlGF no es necesario para el establecimiento y mantenimiento, lo que los autores describieron como “vasos saludables”. Si bien esto puede ser así, el grupo demostró que PlGF, así como VEGF, desempeña un papel importante en la isquemia inducida por angiogenesis. Por lo tanto, el tratamiento con anticuerpos (y especialmente con ambos) puede exacerbar la coexistencia con la patología arterial coronaria o la enfermedad arterial periférica.
Dejando estas advertencias de lado, el peso de la evidencia indica que los macrófagos asociados al tumor juegan un papel importante en la patogénesis de muchos cánceres humanos y sugiere que la terapia anti-PlGF representa una nueva e importante arma en la batalla contra el mal. Así, mientras seguimos avanzando, la combinación de armas a ser utilizadas en cada batalla (es decir, el paciente) para explotar más eficazmente los puntos débiles del enemigo (es decir, el cáncer) puede convertirse en el mayor desafío en la prolongación de la supervivencia de los enfermos.
Fuente bibliográfica
Targeting Tumor Angiogenesis
Gregg L. Semenza, M.D., Ph.D.
Johns Hopkins University School of Medicine, Baltimore, USA.
N Engl J Med. 2008 May 8; 358(19):2066-7