Secuenciación genómica y contaminantes
Esta técnica se usa cada vez más en la investigación de enfermedades infecciosas y microbiomas. Un estudio muestra cómo pueden surgir hallazgos falsos a través de diferentes tipos de contaminación.
La secuenciación del genoma de alto rendimiento está cambiando la forma en que practicamos la medicina al proporcionar nuevas habilidades para identificar las causas genéticas de las enfermedades. Las aplicaciones de secuenciación de ADN se basan con frecuencia en bases de datos seleccionadas para el análisis e interpretación de los resultados. Sin embargo, durante este proceso, es casi imposible evitar pequeñas cantidades de ADN que no pertenecen al organismo de interés. Estos contaminantes provienen de diversas fuentes, incluido el personal de laboratorio, los reactivos utilizados e incluso de las propias muestras. Cuando las muestras bajo investigación son humanas, los contaminantes microbianos pueden interpretarse como agentes infecciosos. Por el contrario, cuando son de bacterias, los contaminantes del genoma humano pueden haberse ensamblado previamente, sin saberlo, en la secuencia de referencia de un genoma bacteriano y, por lo tanto, convertirse en una fuente engañosa cuando tales estructuras se detectan en estudios posteriores.
Un nuevo estudio (Sci Rep 2022; 12:9863-9863) ilustra cómo sucede esto. Se recolectaron las "lecturas" de secuenciación de ADN sin ensamblar de casi 5.000 personas, que se escanearon para identificar virus, bacterias y arqueas. Las que no coincidían con el genoma humano se cotejaron y analizaron para producir una imagen del "contaminoma" humano, que se puede dividir en tres categorías (figura 1A): lecturas virales asociadas con el viroma humano (p. ej., la colección de todos los virus que se encuentran en humanos); lecturas bacterianas o virales introducidas a través de la recolección de muestras (p. ej., microbiota normal del sitio de la muestra) y manipulación, propagación de líneas celulares o reactivos y kits de laboratorio utilizados para la secuenciación (p. ej., contaminantes experimentales); y lecturas bacterianas que coinciden erróneamente debido a la contaminación de la secuencia humana en las bases de datos del genoma bacteriano (es decir, contaminación computacional).
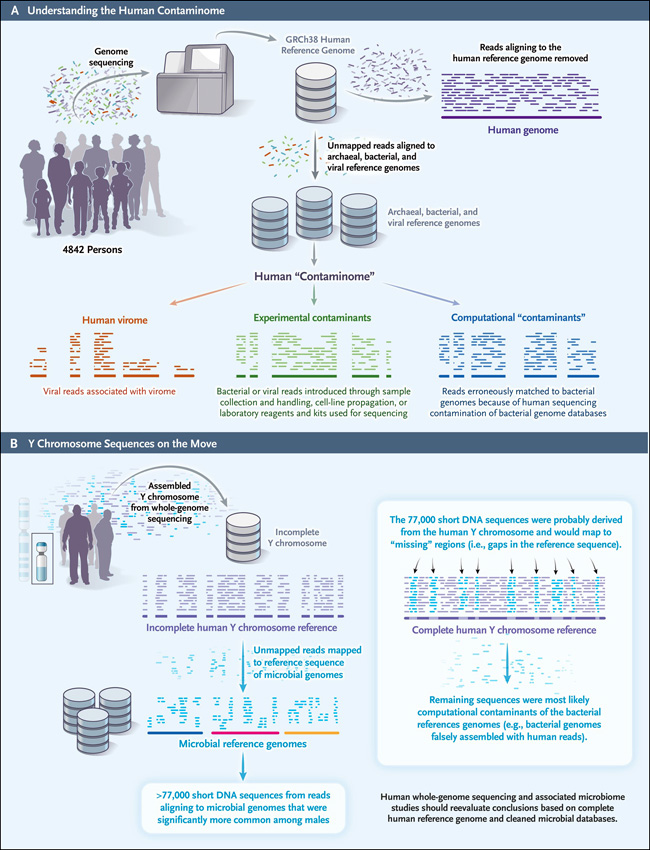
Figura 1: entendiendo el contaminante humano
El peligro de la tercera categoría es que puede conducir a asociaciones espurias entre microbios y enfermedades. Esta investigación lo ilustra con un intrigante hallazgo: después de identificar todas las lecturas bacterianas presentes en las 5.000 muestras de ADN humano, detectaron más de 50 bacterias que eran significativamente más comunes en los hombres versus las mujeres (el sexo se asignó de forma binaria). En lugar de llegar a la rápida conclusión de que estos resultados reflejaban infecciones bacterianas reales que eran más frecuentes en hombres, en su lugar los autores se preguntaron qué sucedería si los genomas bacterianos estuvieran contaminados con fragmentos del cromosoma Y humano (figura 1B) En ese caso, las secuencias derivadas de este cromosoma coincidirían (erróneamente) con las de los genomas bacterianos. El apoyo a esta hipótesis fue el estado de la secuenciación del cromosoma Y, que, hasta principios de este año, todavía estaba inconcluso. Se identificaron 77.647 secuencias cortas de ADN a partir de lecturas que se alinearon con genomas bacterianos que eran significativamente más comunes entre hombres.
Afortunadamente, la secuencia final del cromosoma Y se publicó este año. Se alinearon las 77.647 secuencias "bacterianas" identificadas por el estudio con la secuencia del cromosoma Y, y se observó que 73.691 de ellas (95%) coincidían, indicando que estas secuencias son de hecho humanas, confirmando la hipótesis previa. Este resultado enfatiza la necesidad de ser cauteloso al interpretar los resultados de grandes proyectos de secuenciación de ADN, y plantea la pregunta de si las asociaciones reportadas entre especies microbianas particulares y cáncer, sangre o enfermedades autoinmunes aún se mantienen. ¿Podrían algunos de ellos ser artefactos de contaminantes computacionales?
El problema de la contaminación de la secuencia humana en los genomas bacterianos se extiende más allá del cromosoma Y. Se ha informado que más de 3.000 genomas microbianos contienen pequeños fragmentos humanos. Este complica el uso clínico de genomas microbianos en el diagnóstico de enfermedades infecciosas, en el que la secuenciación de muestras humanas es la base para la identificación de patógenos. Este método implica la comparación de secuencias de ADN o ARN de una muestra de un paciente con las de todos los genomas microbianos conocidos (virus, bacterias, hongos y parásitos) para identificar la causa de la infección. En este contexto, distinguir las lecturas microbianas asociadas con un verdadero patógeno de los contaminantes es esencial para evitar diagnósticos erróneos. A pesar de los mejores esfuerzos de los investigadores, los contaminantes computacionales pueden afectar incluso a las bases de datos más sólidas.
Además de los contaminantes computacionales, los experimentales pueden ser difíciles de discernir, especialmente para muestras con baja biomasa (normalmente tienen una pequeña proporción de ADN microbiano del huésped), como es el caso de la sangre y el líquido cefalorraquídeo. Algunos de los contaminantes experimentales más comunes provienen de patógenos conocidos, incluidos estafilococos, pseudomonas y especies de micobacterias, por nombrar algunos. Afortunadamente, se pueden aplicar controles experimentales bien diseñados para detectarlos estos, siempre que se sepa que existen.
Los resultados informados ilustran cómo cada proyecto de secuenciación del genoma humano captura diferentes formas de vida, incluidas secuencias de ADN de bacterias y virus, y cómo podrían surgir asociaciones falsas entre enfermedades infecciosas y rasgos (como el sexo). Este trabajo enfatiza la obligación de tener secuencias genómicas completas y precisas para evitar la contaminación computacional de las secuencias de referencia y mejorar la precisión del diagnóstico. También subraya la necesidad de protocolos estándar para identificar el "contaminoma", a fin de garantizar la fidelidad de los estudios y diagnósticos basados en secuenciación.
Fuente bibliográfica
The Human “Contaminome” and Understanding Infectious Disease
Patricia J. Simner, Ph.D., and Steven L. Salzberg, Ph.D.
Department of Pathology, Division of Medical Microbiology (P.J.S.), the Department of Medicine, Division of Infectious Diseases (P.J.S.), and the Department of Biomedical Engineering (S.L.S.), Johns Hopkins School of Medicine, the Department of Computer Science and Center for Computational Biology, Whiting School of Engineering (S.L.S.), and the Department of Biostatistics, Bloomberg School of Public Health (S.L.S.), Johns Hopkins University, Baltimore.
N Engl J Med 2022; 387:943-946