Mutaciones silenciosas: ni tan neutrales
Los genomas tienen diferentes tipos de variaciones genéticas, y se supone que muchas son "silenciosas", es decir, no tienen efecto sobre la biología, la salud o el estado físico. Un reciente estudio muestra que esto no es tan así.
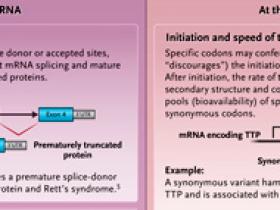
Una creencia frecuente es que los cambios de codones de carácter sinónimo, llamados así porque no alteran la codificación de aminoácidos, son variantes genéticas neutras que no tienen efecto sobre el fenotipo o la aptitud genética. Sin embargo, una creciente literatura observa que sí influirían en la biosíntesis proteica y el comportamiento biológico de las células y subyacen a la enfermedad humana. Por ejemplo, impactan en la susceptibilidad a la enfermedad de Crohn, promueven el plegamiento incorrecto de la forma mutante más común de la proteína de regulación de la conductancia transmembrana de la fibrosis quística (CFTR) en la fibrosis quística y alteran la especificidad de las bombas de salida de fármacos en las células cancerosas. Se sabe que las variantes sinónimas asociadas a la enfermedad afectan una variedad de procesos celulares que abarcan el ARN mensajero (ARNm) y la síntesis de proteínas (figura 1). Sin embargo, persiste la creencia en su neutralidad general, lo que tiene ramificaciones para la genética de poblaciones y la interpretación de los resultados genómicos en la clínica. El flujo gradual de evidencia que describe variantes sinónimas funcionales hace que sea difícil saber con qué frecuencia ocurren y el alcance de sus efectos en los procesos biológicos humanos.
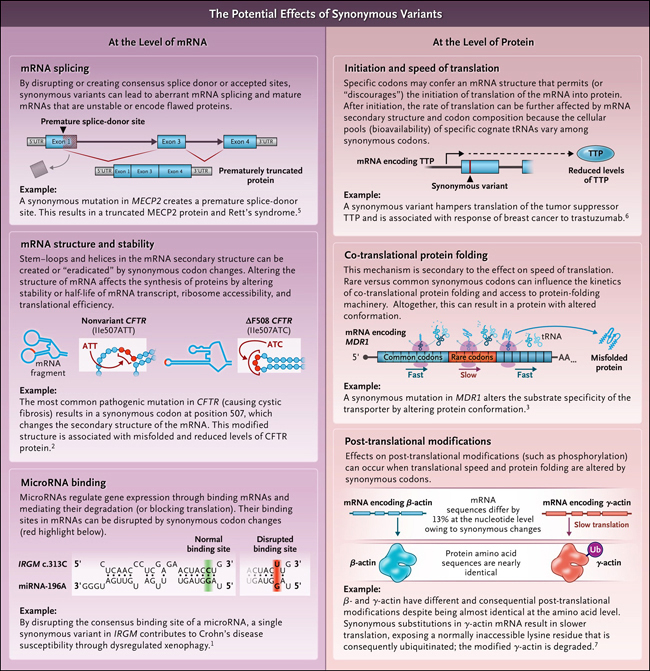
Figura 1: los efectos potenciales de las variantes sinónimas.
¿Se puede probar experimentalmente y a escala la no neutralidad de las mutaciones sinónimas? Xukang Shen y colegas (Nature 2022; 606:725-731) abordaron esta pregunta utilizando la levadura Saccharomyces cerevisiae (figura 2). El trabajo fue posible gracias a los avances metodológicos mediante una herramienta de edición de genes llamada CRISPR-Cas9 (abreviatura de "repeticiones palindrómicas cortas agrupadas regularmente interespaciadas asociada a la proteína 9"), que permitió la creación sistemática de mutaciones específicas sinónimas y no sinónimas. Los autores crearon más de 8.000 variantes de un solo nucleótido en S. cerevisiae dentro de las regiones codificantes en 21 genes no esenciales. A continuación, se evaluó cada variante en cuanto a su efecto sobre la aptitud de S. cerevisiae. La medición de la aptitud relativa se logró cultivando los mutantes con levadura de tipo salvaje y controlando los cambios en las frecuencias del genotipo (que reflejan las modificaciones relativas en la levadura mutante frente a la de tipo salvaje) a lo largo del tiempo. Con esto, se puede medir de forma fiable pequeñas diferencias absolutas en la aptitud entre variantes (figura 2).
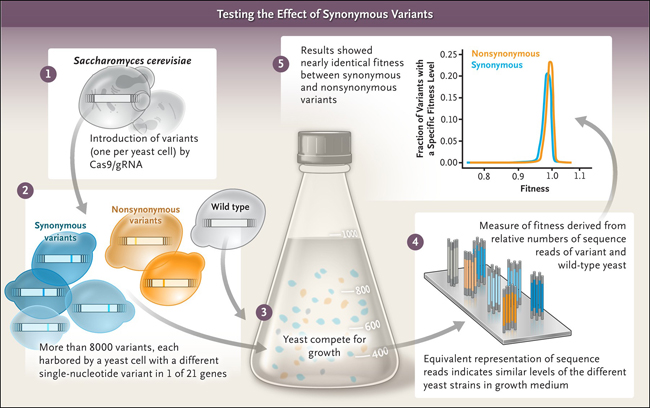
Figura 2: prueba del efecto de variantes sinónimas.
La aptitud mediana de los mutantes con mutaciones sin sentido (que anulan la función de la proteína) fue, como se esperaba, más baja que la de las variantes no sinónimas. Sorprendentemente, tres cuartas partes de las primeras mostraron una aptitud reducida, y la mediana de la aptitud para 1.866 variantes fue casi idéntica a la de las 6.306 no sinónimas. Este sorprendente resultado contradice directamente la neutralidad de las mutaciones sinónimas que se han mantenido durante décadas. A continuación, se buscaron mecanismos comunes subyacentes a los cambios observados en la forma física, descubriéndose que la aptitud reducida para las variantes sinónimas y no sinónimas se correlacionaba más estrechamente con la reducción relativa a nivel de expresión del ARNm. Este es un hallazgo algo inesperado porque se creía que las variantes no sinónimas no ejercían efectos sobre los niveles de ARNm.
Estos resultados conllevan una serie de implicaciones. Además de desafiar los supuestos de la genética de poblaciones, también cuestionan el enfoque analítico estándar para la secuenciación clínica. Las pruebas genómicas han brindado a los médicos una capacidad sin precedentes para comprender la base genética de las enfermedades humanas, detectar mutaciones que provocan cáncer, diagnosticar síndromes familiares y justificar el uso de terapias dirigidas. Los profesionales están acostumbrados a recibir un informe de secuenciación con una lista de variantes categorizadas en una escala de probabilidad patogénica.
Sin embargo, un aspecto pasado por alto de la genética clínica es el flujo de trabajo bioinformático para identificar y atribuir patogenicidad (o un estado benigno) a las variantes genéticas. La secuenciación del exoma y genoma genera millones de variantes que deben filtrarse hasta las que sean médicamente significativas. Las pautas del Colegio Estadounidense de Genética Médica y Genómica establecen que las variantes sinónimas, a menos que interrumpan el empalme, deben clasificarse como "probablemente benignas". Ciertamente, nuestra capacidad para evaluarlas mejor dependerá del desarrollo de herramientas computacionales confiables para considerarlas integralmente. Tales herramientas podrían tener en cuenta el contexto de la secuencia de la variante y el conocimiento sobre cómo podría afectar la interacción entre el ARNm y el ribosoma durante la síntesis de proteínas.
El presente trabajo también se relaciona con el desarrollo de proteínas terapéuticas recombinantes. La mayoría son producto de la ingeniería. La modificación de la proteína nativa permite una función mejorada o una vida media prolongada. La secuencia de su expresión se puede revisar con sustituciones de codones sinónimos para facilitar la clonación o lograr una mayor expresión de la proteína en una célula huésped. La capacidad para modular proteínas terapéuticas se ha expandido rápidamente, pero predecir cuándo y cómo las sustituciones sinónimas afectan la estructura proteica y el riesgo de inmunogenicidad sigue siendo un desafío. Experimentos a gran escala deberían motivar más estudios que evalúen cómo las variantes sinónimas influyen en la expresión y función de las proteínas, y en qué medida son relevantes para la enfermedad humana.
Fuente bibliográfica
When Silence Disrupts
R.C. Hunt and C. Kimchi-Sarfaty
Division of Plasma Protein Therapeutics, Office of Tissues and Advanced Therapies, Center for Biologics Evaluation and Research, Food and Drug Administration, Silver Spring, MD.
N Engl J Med 2022; 387:753-756