Mitofagia, inflamación y párkinson
Las afecciones neurodegenerativas, como las enfermedades de Parkinson y Alzheimer, constituyen una importante carga para la salud. Aunque los síntomas, o las células afectadas, pueden diferir en tales trastornos, algunas patologías neurodegenerativas tienen ciertas características en común. Por ejemplo, un estado de inflamación y una eliminación alterada de organelos defectuosos. Sin embargo, queda por determinar si estas alteraciones compartidas están interconectadas y si son una causa o una consecuencia de la enfermedad. En un reciente estudio, se dan a conocer hallazgos en un modelo animal con alteraciones genéticas relacionados con la enfermedad de Parkinson. Los autores identifican una conexión directa entre el proceso celular que elimina la mitocondria dañada - llamada mitofagia - y la inflamación.
Proteína STING e inflamación
Las enzimas PINK1 y parkin actúan en una vía que une una proteína llamada ubiquitina a proteínas celulares; estos componentes marcados con ubiquitina son el objetivo de la destrucción celular. Estas enzimas ayudan en el proceso de mitofagia, en el cual los fragmentos mitocondriales no funcionales son rápidamente secuestrados en una vesícula unida a la membrana que se degrada cuando se fusiona con lisosomas.
Las mutaciones que impiden la expresión normal de PINK1 o parkin están relacionadas con una forma de aparición temprana de la enfermedad de Parkinson, y existen pruebas de que si no se eliminan con éxito las mitocondrias dañadas, el riesgo de desarrollar la enfermedad es mayor. Sin embargo, ratones deficientes en PINK1 o parkin no desarrollan síntomas del tipo observado en personas con anomalías en la expresión de estas proteínas; tales síntomas incluyen problemas de movimiento derivados de la pérdida de células neuronales que producen dopamina. Estos animales tampoco tienen el alto nivel de inflamación que caracteriza a la enfermedad de Parkinson.
El hallazgo de que la pérdida de PINK1 o parkin tiene un efecto mínimo en los animales fue sorprendente, porque durante mucho tiempo se pensó que la remoción de las mitocondrias dañadas tiene un papel clave en la protección de las células contra el daño oxidativo. Las mitocondrias defectuosas representan una amenaza grave para las células porque pueden liberar especies reactivas de oxígeno (ROS) que causan daños celulares sustanciales. Por ejemplo, las ROS pueden aumentar la carga de agregados proteicos potencialmente tóxicos si las proteínas están sujetas a daños causados por ROS. Las mitocondrias defectuosas también pueden liberar componentes que normalmente no están presentes en el citoplasma, como el ADN mitocondrial. De hecho, la infiltración de ADN mitocondrial en el citoplasma puede desencadenar una respuesta inflamatoria mediada por la proteína STING. Esto plantea la pregunta de si la protección contra la inflamación, más que contra el daño oxidativo, podría ser el papel clave de la mitofagia en el contexto de la enfermedad de Parkinson.
Danielle A. Sliter y colaboradores investigaron las consecuencias de la pérdida de PINK1 o parkin en ratones que estaban sujetos a un alto nivel de estrés mitocondrial (DOI: 10.1038/s41586-018-0448-9). Este estrés se produjo al someter a los animales a un régimen de ejercicio intensivo y agotador o al explotar una alteración genética que se encuentra en ratones mutantes, en los que una enzima polimerasa defectuosa causa un alto nivel de mutaciones en el ADN mitocondrial. Previamente se conocía que los ratones mutantes viejos que carecen de parkin tienen menos neuronas secretoras de dopamina, y que estos ratones desarrollan anormalidades de movimiento que se asemejan a las observadas en personas que tienen la enfermedad de Parkinson. Cuando los autores impusieron el estrés mitocondrial a los animales que carecían de PINK1 o parkin, descubrieron que el nivel sanguíneo de citoquinas proinflamatorias era mucho más alto que en los ratones que no estaban sujetos a este estrés mitocondrial.
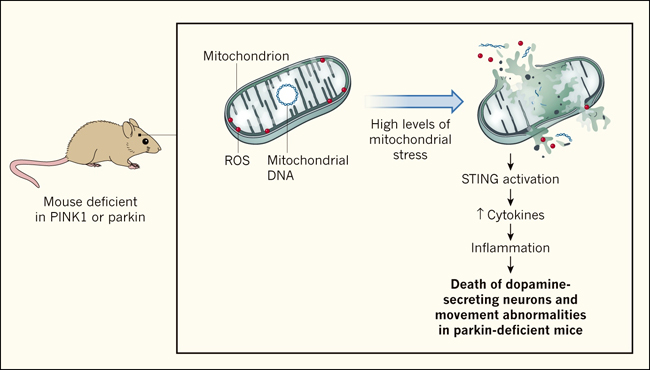
Figura 1. La ausencia de las proteínas PINK1 o parkin conduce a la inflamación.
Las anomalías en las proteínas PINK1 o parkin están relacionadas con la enfermedad de Parkinson de inicio temprano en humanos. Los ratones que carecen de cualquiera de las dos proteínas son deficientes en el proceso de eliminación controlada de los mitocondrias dañadas.Este mecanismo es necesario para prevenir la ruptura de los organelos con la posterior liberación de especies reactivas de oxígeno (ROS) y ADN mitocondrial en el citoplasma. Sin embargo, estos animales no tienen los tipos de síntomas que se encuentran en la enfermedad de Parkinson en humanos. Sliter y sus colegas (DOI: 10.1038/s41586-018-0448-9) indujeron altos niveles de estrés mitocondrial en estos ratones (por el uso de niveles excesivos de ejercicio o por un alto nivel de mutaciones de ADN mitocondrial) y descubrieron que la activación de la proteína STING -que puede mediar la inflamación cuando el ADN mitocondrial entra en el citoplasma- aumenta la expresión de citoquinas que inducen inflamación. Esto indica que el PINK1 y parkin protegen contra la inflamación, lo que podría explicar la inflamación observada comúnmente en personas con la enfermedad de Parkinson. En ratones viejos que carecen de parkin, la inflamación mediada por STING se correlaciona con anormalidades en el movimiento y pérdida de células neuronales secretoras de dopamina.
Sin embargo, los autores encontraron que si los ratones carecían de STING, así como de PINK1 o parkin, la expresión de citocinas inflamatorias no aumentaba como resultado del estrés mitocondrial. Esto indica que STING es necesaria para impulsar la inflamación mediada por este tipo de estrés (figura 1). Además, la ausencia de STING evitó los defectos de movimiento y las pérdidas neuronales que suelen ocurrir en ratones mutantes viejos que carecen de parkin. Los autores encontraron que los niveles sanguíneos de citocinas inflamatorias IL-6, IL-1β y CCL2, que se elevan por encima de lo normal en estos mismos ejemplares, también son más altos de lo normal en personas con enfermedad de Parkinson que tienen mutaciones en ambas copias del gen parkin. Sin embargo, los autores observaron que estas citoquinas también estaban elevadas en parientes libres de enfermedad de personas que tienen la enfermedad de Parkinson. El estudio de estos parientes, que tienen una mutación en sólo una de sus dos copias del gen parkin, sugiere que estas alteraciones particulares de citoquinas no son suficientes para causar la enfermedad. Curiosamente, las personas que reciben tratamiento a largo plazo con medicamentos antiinflamatorios no esteroidales tienen un riesgo inferior al promedio de desarrollar la enfermedad de Parkinson. Esta observación es consistente con un modelo en el que los bajos niveles de inflamación podrían proteger contra la neurodegeneración.
Los resultados plantean muchas preguntas importantes. ¿Cómo la inflamación mediada por STING es capaz de causar la muerte neuronal? ¿Por qué están específicamente afectadas las células neuronales secretoras de dopamina? ¿La inflamación dependiente de STING está relacionada con otras anomalías asociadas con la neurodegeneración, como la formación de agregados de proteínas?
Sin embargo, antes de que estas preguntas puedan ser respondidas en el contexto de la enfermedad humana, una consideración crucial es qué tan bien estos ratones proporcionan un modelo de la enfermedad de Parkinson en humanos. Otros conocimientos podrían provenir del uso de otros sistemas, como ratas o moscas de la fruta (Drosophila), que imitan mejor los tipos de cambio que ocurren en el párkinson. Finalmente, dado que la mitofagia dañada y la inflamación son características comunes de varios trastornos neurodegenerativos, es tentador especular que la inflamación dependiente de STING podría contribuir de manera similar a otras condiciones neurodegenerativas, como la enfermedad de Alzheimer. Sería interesante probar esta hipótesis experimentalmente.
Fuente bibliográfica
Elusive inflammation insight uncovered
Alexandra Stolz and Ivan Dikic
Institute of Biochemistry II and Buchmann Institute for Molecular Life Sciences, Goethe University School of Medicine, 60590 Frankfurt am Main, Germany.
DOI: 10.1038/d41586-018-05988-z