La vía rápida de la aterosclerosis
El envejecimiento se asocia con un aumento en la frecuencia de mutaciones somáticas en células hematopoyéticas. Varias de estas recurrentes alteraciones que incluyen las acontecidas en genes codificantes para enzimas modificadoras epigenéticas como TET2, promueven la expansión de células sanguíneas anómalas. Esta hematopoyesis clonal se correlaciona con un mayor riesgo de enfermedad cardiovascular aterosclerótica. En recientes estudios se dilucidaron los efectos de la expansión de células mutantes TET2 en ratones propensos a la aterosclerosis que carecen del receptor de la lipoproteína de baja densidad. Se observó que la reconstitución de la médula ósea con células deficientes en TET2 era suficiente para su expansión clonal potenciando el incremento del tamaño de placas ateroscleróticas. Los macrófagos TET2 deficientes exhibieron un aumento en la secreción de interleuquina-1β y del complejo inflamosoma lo que apoya la hipótesis de que las mutaciones somáticas en TET2 juegan un papel causal en la aterosclerosis.
Macrófagos mutantes
El envejecimiento es un importante factor de riesgo para una condición llamada aterosclerosis, en la que el colesterol se acumula en las arterias en forma de placas. Cuando las placas sufren inflamación, se pueden romper o erosionar, lo que conduce a la formación de coágulos de sangre que ocluyen las arterias y causan ataques cardíacos y accidentes cerebrovasculares (ACV). Un posible factor causal es la hematopoyesis clonal - un fenómeno en que surgen mutaciones en células troncales hematopoyéticas formadoras de la sangre (HSC), evento producido durante el envejecimiento y que promueve la proliferación de poblaciones de células sanguíneas portadoras de estas mutaciones a expensas de linajes celulares de tipo silvestre. Sin embargo, la manera en que este fenómeno podría causar aterosclerosis no se ha dilucidado. En un reciente artículo (Science. 2017 Feb 24;355(6327):842-847) Fuster y sus colegas esbozaron una vía por la que la deficiencia del gen Tet2 puede originar aceleradamente la patología a través de hematopoyesis clonal en ratones.
En 2014, un estudio (N Engl J Med. 2014 Dec 25;371(26):2488-98) reveló que las mutaciones que causan la hematopoyesis clonal ocurren en más de un 10% de las personas mayores de 70 años. Tales mutaciones eran más comunes en genes como TET2, el que codifica para proteínas que modulan la adición o eliminación de modificaciones moleculares en el ADN para alterar la expresión génica. Otros genes frecuentes donde ocurrían, era en genes que codifican proteínas reguladores de señalización celular. Estas anomalías promueven la auto-renovación de las HSC y la proliferación de monocitos y neutrófilos portadores de mutaciones, los que surgen a partir de sus precursoras HSC. El estudio encontró una fuerte asociación entre mutaciones y un aumento del riesgo de cáncer de la sangre. Pero las mutaciones también estaban relacionadas con una mayor mortalidad general, lo que refleja en gran medida un elemento aumento de ataques cardíacos y ACV.
La actividad de la proteína TET2 conduce a la eliminación de grupos metilo a partir de una forma modificada de la base citosina del ADN. Por lo tanto, la deficiencia de TET2 resulta en el aumento de la metilación del ADN, lo que altera la expresión de un espectro de genes implicados en el desarrollo de tumores. Fuster y sus colegas investigaron el papel de esta proteína en la aterosclerosis mediante el uso de un modelo de ratón de hematopoyesis clonal. Mezclaron las células de médula ósea que carecen de Tet2 y células de tipo salvaje en una proporción de 10% a 90%, para imitar la configuración en la que ocurre la hematopoyesis clonal en personas de edad avanzada. Luego, los autores trasplantaron estas células en ratones que habían sido genéticamente diseñados para ser propensos a la aterosclerosis y que habían sido Irradiados por lo que carecían de su propias células de la médula ósea.
Como se encontró en un estudio anterior (Blood. 2011 Oct 27;118(17):4509-18), las células deficientes en Tet2 eran mucho más abundantes que las células de tipo silvestre en la médula ósea y la sangre de estos ratones, lo que indica que la hematopoyesis clonal estaba ocurriendo (Figura 1). A pesar de un aumento de la auto-renovación de las HSC, el total de células blancas de la sangre fueron normales, como se ve en ratones y humanos que portan mutaciones en Tet2. Sin embargo, animales de trasplante mixto exhiben aterosclerosis acelerada en comparación con ratones que solamente habían recibido células de tipo silvestre.
La hipótesis de Fuster y sus colegas propuso que esta aceleración podría ser el resultado de alteraciones en un subconjunto específico de células inmunes llamadas macrófagos. Estas células, derivadas a partir de monocitos, incorporan formas modificadas de lipoproteínas de baja densidad y promueven inflamación en arterias ateroscleróticas. Para probar su teoría, los autores eliminaron Tet2 específicamente en los macrófagos, y esto de nuevo condujo a una aterosclerosis acelerada. En concordancia con trabajos previos, cuando los investigadores cultivaron macrófagos deficientes en Tet2 in vitro, observaron una sobrerregulación de muchas proteínas proinflamatorias secretadas. Sin embargo, la examinación in vivo de las placas sugirió un rol predominante para una clase de estas proteínas: interleuquina 1β (IL-1β).
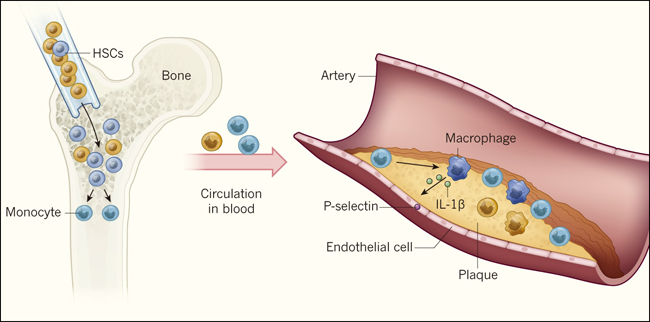
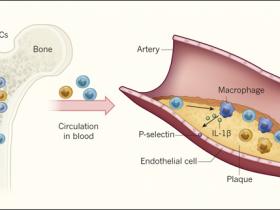
Figura 1: un mecanismo para la aterosclerosis acelerada.
Fuster y sus colegas investigaron cómo la aterosclerosis - acumulación de placas ricas en colesterol en los vasos sanguíneos - podría ser promovida por mutaciones en el gen Tet2 en células madre hematopoyéticas (HSC). Los investigadores trasplantaron médula ósea que contenía una mezcla de HSC Tet2 deficientes (azul) y HSC de tipo silvestre (naranja) en ratones. La proliferación de HSC mutantes y de monocitos derivados de HSC conlleva a un mayor crecimiento de células mutantes respecto a células de tipo salvaje en la médula ósea y la sangre. En las arterias, los monocitos son reclutados hacia las placas, donde dan lugar a macrófagos que causan inflamación. Un complejo llamado inflamasoma (no mostrado) en los macrófagos, escinde y activa la proteína IL-1β, la que es secretada e induce la expresión de la proteína P-selectina en células endoteliales que revisten los vasos sanguíneos. Esto aumenta el reclutamiento de monocitos a las placas dando lugar a más macrófagos proinflamatorios, acelerando la aterosclerosis.
IL-1β se produce inicialmente como una proteína precursora que se activa a través de escisión por la caspasa 1 en una estructura subcelular llamada inflamasoma. La activación del inflamasoma es un proceso de dos pasos: en el primero, hay un aumento de la producción de de ARN mensajero de IL-1β y de genes que codifican componentes del inflamasoma; esto es seguido por una segunda señal que induce la activación del inflamasoma. Fuster y sus colegas mostraron que en la fase inicial se daba un incremento de macrófagos deficientes en Tet2 en comparación con los controles. Además, cuando los autores trataron las células con ATP o con cristales de colesterol (segunda señal para la activación del inflamasoma que probablemente se presenta durante la aterosclerosis), se observó un aumento de la activación de caspasa 1 y de secreción de IL-1β.
De acuerdo con estos hallazgos, los niveles de una molécula cuya expresión está regulada por IL-1β - la proteína P-selectina - eran elevados en células endoteliales (que bordean las paredes de las arterias) en ratones sometidos a un trasplante mixto en comparado con controles de tipo salvaje. La P-selectina recluta monocitos hacia placas ateroscleróticas dando origen a más macrófagos, aumentando la inflamación y la eventual ruptura de la placa. Fuster y sus colegas demostraron que el reclutamiento de monocitos fue mayor en los ratones que carecían de Tet2 en macrófagos. Por último, los autores demostraron que un inhibidor del inflamasoma bloquea el aumento de la secreción de IL-1β y de la aterosclerosis en ratones deficientes en Tet2. En conjunto, estos datos demuestran que el desarrollo de la aterosclerosis se acelera por mutaciones en Tet2 que conducen a una hematopoyesis clonal y a la activación de inflamosomas IL-1ß en los macrófagos.
Fuente bibliográfica
Cardiovascular disease: Commonality with cancer
Alan R. Tall & Ross L . Levine
Division of Molecular Medicine, Department of Medicine, Columbia University, New York.
doi:10.1038/nature21505