Inteligencia artificial y biomedicina
En cuestión de meses, la aplicación de la inteligencia artificial y aprendizaje automático a la predicción de estructuras proteicas tridimensionales ha generado una explosión de conocimientos que promete un buen augurio para acelerar la identificación y selección de proteínas "farmacológicas".
La predicción de la estructura de las proteínas con el uso de inteligencia artificial y aprendizaje automático (AI-ML) está preparada para transformar cómo y cuándo se utiliza la información de bioestructura tridimensional (3D) en investigación y práctica clínica. Hasta hace poco, el conocimiento detallado de las estructuras 3D de macromoléculas biológicas estaba restringido a aproximadamente unas 180.000 estructuras a nivel atómico determinadas experimentalmente y archivadas en el Protein Data Bank (PDB) global (ver www.pdb.org), así como sus homólogos cercanos o parálogos. El PDB se fundó en 1971 como el primer recurso de datos digitales de acceso abierto conocido en biología. La información almacenada ha informado la comprensión de los mecanismos de la enfermedad, la identificación de dianas terapéuticas, el descubrimiento de fármacos de moléculas pequeñas y el diseño de productos biológicos.
Hacia fines de 2020, Google DeepMind anunció que su sistema computacional AlphaFold2 AI-ML podría modelar estructuras 3D de pequeñas proteínas globulares con precisiones similares a las obtenidas a través de los experimentos. Se ha publicado el método DeepMind, y los modelos de estructura (o AI-ML) en 21 proteomas (incluido uno humano, así como los proteomas de otros organismos y de algunos cultivos agrícolas y patógenos), los que están disponibles gratuitamente. DeepMind no está solo; también RoseTTAFold proporciona el modelado de la estructura de proteínas y acceso al código informático. En cuestión de meses, el conocimiento de las bioestructuras 3D pasó de la cobertura parcial del proteoma humano y un número limitado de otros proteomas a modelos de estructura AI-ML razonablemente precisos. Esta explosión de información 3D transformará la biomedicina.
Sin embargo, los modelos de estructura AI-ML no desplazarán los datos PDB. Los modelos y los datos de PDB son más valiosos cuando se usan juntos, como se ilustra mediante una comparación de los flujos de trabajo y los resultados para la determinación de la estructura experimental y el modelado de la estructura AI-ML del antígeno ventral neuro-oncológico 2 de la proteína de unión al ARN humano (Nova-2) (figura 1). Nova-2 está involucrado en trastornos del neurodesarrollo, autismo, polineuropatías paraneoplásicas y síndrome de X frágil. Una estructura PDB del tercer dominio de homología K (KH3) de Nova-2 más una horquilla de ARN (PDB ID, 1ec6) reveló un reconocimiento de ácido nucleico específico de secuencia a nivel atómico (figura 1, panel izquierdo). Las estructuras de PDB estrechamente relacionadas están restringidas a los otros dos dominios de homología K de Nova-2 humana y los tres dominios de homología K de su parálogo, Nova-1. Por el contrario, el modelo de estructura humano Nova-2 AI-ML abarca la totalidad de esta proteína multidominio, asemejándose a "espaguetis y albóndigas" (figura 1, panel derecho). De manera tranquilizadora, las predicciones AlphaFold2 de alta confianza para los tres dominios de homología de Nova-2 K se parecen mucho a sus estructuras PDB. Son las "albóndigas". Los segmentos restantes de la cadena polipeptídica se parecen a los "espaguetis". Carecen de una estructura definida y es casi seguro que estén desordenados. AlphaFold2 los designa como de confianza baja a muy baja, y sus coordenadas atómicas calculadas no son precisas y no deben tomarse literalmente. En particular, la comparación lado a lado de la estructura PDB y el modelo AI-ML revela cómo el dominio KH3 de Nova-2, en el contexto de la proteína de longitud completa, se une al ARN.
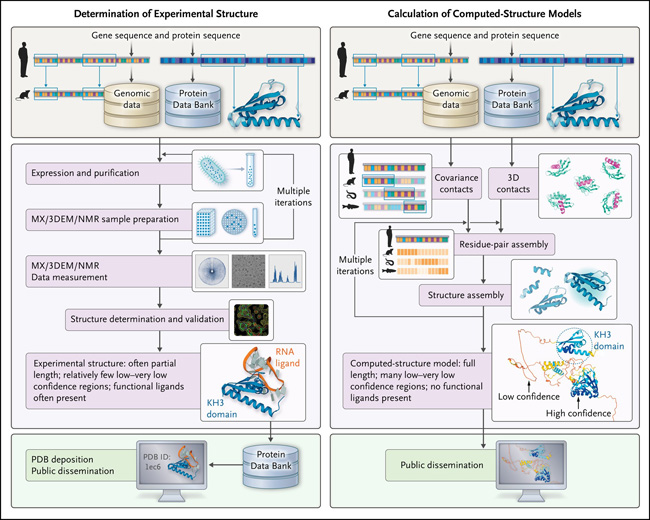
Figura 1: flujos para la determinación de estructura experimental versus flujos para el cálculo de modelos de estructura computacional.
Se muestra una comparación de los resultados de dos enfoques para obtener información sobre una estructura tridimensional para el antígeno ventral-2 neuro-oncológico de la proteína de unión al ARN humano (Nova-2). Interacciones entre aminoácidos inferidos de la variación evolutiva. 3DEM denota microscopía electrónica, homología de tercer K de KH3, cristalografía macromolecular MX y NMR espectroscopía de resonancia magnética nuclear.
Además de contribuir al conocimiento de la estructura 3D a través de proteomas completos (similar a "listas de partes" para biología y biomedicina), el preciso modelado de la estructura computacional permitirá el análisis de cambios genéticos clínicamente relevantes manifestados en tres dimensiones por proteínas individuales. Por ejemplo, la variante B.1.617.2 (delta) del síndrome respiratorio agudo severo coronavirus 2 (SARS-CoV-2) porta 13 mutaciones sin sentido (que efectúan cambios de aminoácidos) y una deleción de dos residuos. Aunque el PDB alberga actualmente más de 1500 estructuras experimentales relacionadas con la enfermedad por coronavirus 2019 (Covid-19), incluidas más de 500 estructuras relacionadas con la proteína spike [S], actualmente no hay estructuras de la variante delta S de longitud completa. proteína en el archivo. La figura 2A representa un modelo de estructura computarizada de la proteína S de la variante delta que muestra las 13 sustituciones de aminoácidos, incluido un cambio crítico en el residuo 452 que mejoraría la unión de spike a su receptor celular (enzima convertidora de angiotensina 2). Cuando se detecte otra variante preocupante del SARS-CoV-2 (o un nuevo coronavirus patógeno) y se secuencie su genoma, el modelado de la estructura computacional será invaluable en el diseño de nuevas vacunas y en el descubrimiento de anticuerpos monoclonales terapéuticos-diagnósticos o pequeños. -Moléculas que se dirigen a proteínas virales esenciales.
El acceso abierto a los modelos de estructura AI-ML también afectará directamente la atención del paciente, complementando los enfoques existentes centrados en la información de la secuencia del genoma y la proteína. Se prevé que la mayoría de las aplicaciones a corto plazo del modelado de estructuras computacionales se centrarán en mutaciones puntuales detectadas en tumores humanos (de línea germinal o somática). Predecir genes frecuentemente mutados que codifican oncoproteínas, como el receptor del factor de crecimiento epidérmico en cánceres de pulmón 6 (figura 2B), ayudará a diferenciar las verdaderas variantes que provocan cáncer de las que no tienen relevancia clínica, para permitir el descubrimiento de terapias, explicar la resistencia a los fármacos e informar los planes de tratamiento. Cualquiera que sea el área terapéutica, la sinergia entre los datos PDB y los modelos de estructura AI-ML acelerará la transición de los programas de investigación biomédica desde una base de datos y el computador.
El uso productivo de esta nueva riqueza de información de bioestructura 3D podría verse obstaculizado por la falta de coherencia o reproducibilidad con métodos de laboratorio que a menudo obstaculizan la aplicación clínica. Aún no se ha llegado a un consenso sobre cómo las características estructurales de una proteína influyen en la actividad in vivo. La aplicación de enfoques basados en IA-ML en este campo podría producir una mejor comprensión y (en última instancia) una predicción de las formas en que las estructuras de proteínas en 3D dictan la función biológica o bioquímica. En el futuro, los hitos clave incluirán una sólida predicción de la selectividad y afinidad de la unión, eficacia en el objetivo, y estudios preclínicos y clínicos de fase inicial respecto a la toxicidad de anticuerpos, peptidomiméticos y de otros agentes moleculares. Finalmente, hay que descubrir y apuntar a proteínas que se puedan administrar como fármacos mediante el uso de métodos que sean ampliamente aplicables en el campo de la medicina.
Fuente bibliográfica
Predicting Proteome-Scale Protein Structure with Artificial Intelligence
S.K. Burley, W. Arap, and R. Pasqualini
Research Collaboratory for Structural Bioinformatics Protein Data Bank, the Institute for Quantitative Biomedicine, and the Department of Chemistry and Chemical Biology, Rutgers, the State University of New Jersey (S.K.B.), and the Rutgers Cancer Institute of New Jersey, New Brunswick (S.K.B.) and Newark (W.A., R.P.); and the Division of Hematology–Oncology, Department of Medicine (W.A.), and the Division of Cancer Biology, Department of Radiation Oncology (R.P.), Rutgers New Jersey Medical School, Newark; and the Research Collaboratory for Structural Bioinformatics Protein Data Bank, San Diego Supercomputer Center, University of California, San Diego, San Diego (S.K.B.).
N Engl J Med 2021; 385:2191-2194