Entendiendo el síndrome de Usher
El síndrome de Usher es un trastorno genético que implica la pérdida auditiva neurosensorial y retinitis pigmentosa. Aunque se le considera una enfermedad rara, es la causa más frecuente de ceguera-sordera en humanos. Aproximadamente de 3 a 6 por ciento de todos los niños sordos y otro 3 a 6 por ciento de los que tienen problemas graves de audición padecen el trastorno. En países desarrollados, como Estados Unidos, alrededor de cuatro recién nacidos por cada 100.000 tienen el síndrome.
Actualmente, no existe una cura posible. La mejor terapia consiste en la identificación temprana para que los programas educativos puedan comenzar tan pronto sea posible. La naturaleza exacta de estos esquemas dependerá de la severidad de la pérdida de audición y visión, así como la edad y las capacidades personales. Normalmente, el tratamiento incluye audífonos, dispositivos de asistencia auditiva, implantes cocleares u otros métodos de comunicación, como lenguaje de señas, orientación y entrenamiento de movilidad.
Recuperación auditiva en el síndrome de Usher
Los intentos de restaurar o prevenir la pérdida de audición con base genética por medio de la intervención biológica no han tenido mayor éxito, principalmente debido a las limitaciones inherentes de la perdida de células mecano-sensoriales y el rescate funcional de las células dañadas. Aunque el tratamiento se basa principalmente en el uso de implantes cocleares (el Premio de Investigación Médica de la Clínica Lasker-DeBakey de 2013 fue otorgado a los pioneros de esta tecnología), éstos están aún lejos de ser lo ideal. La elucidación de la base genética y los avances en la entrega de genes de audición puede cambiar esta imagen. A principios de este año, Jennifer J. Lentz y colaboradores (Nat Med 2013; 19:345-350) reportaron la recuperación auditiva de baja y media frecuencia y la función vestibular en un modelo murino para el síndrome de Usher mediante el uso de oligonucleótidos antisentido.
El síndrome de Usher incluye un grupo de trastornos recesivos caracterizados por deficiencia auditiva, ceguera resultante de la retinitis pigmentosa, y la presencia o ausencia de disfunción vestibular. Se han descrito tres tipos (1, 2 y 3), con al menos 10 genes involucrados; una mutación en cualquiera de éstos es suficiente para promover la enfermedad.
Estos genes codifican proteínas para las células pilosas del oído interno, que traducen las oscilaciones mecánicas de las estructuras cocleares en impulsos neuronales en el nervio auditivo. Dos de estas proteínas, cadherina 23 y protocadherina 15, forman las uniones de punta de los estereocilios, proyecciones que se extienden desde la superficie de la célula del pelo y que son desviadas por las vibraciones cocleares. Los extremos de enlaces de las puntas se unen a los estereocilios unos a otros en filas y se estiran, de tal manera que éstos abren y cierran los canales de cationes adyacentes a las uniones de punta. El resultado de esta acción es la liberación de neurotransmisores que interactúan con los terminales del nervio auditivo, haciendo que se activen. Se sabe que la proteína harmonina, un componente del complejo de las proteínas que anclan las uniones de punta, regula canales de calcio Cav1.3 y la exocitosis.
Los pacientes con mutaciones en USH1C, que codifica la harmonina, padecen el síndrome de Usher tipo 1, la forma más grave de la condición. Se caracterizan por una profunda pérdida de audición y la ausencia de la función vestibular, desde el nacimiento, y la retinitis pigmentosa aparece en los primeros años de la adolescencia. Aunque las estimaciones indican que el síndrome se produce en todo el mundo, con una prevalencia de 3 a 6 casos por cada 100.000 personas, hasta ahora se han reportado portadores de USH1C mutante sólo en la población Acadiana de Louisiana y en familias paquistaníes y libanesas. El alelo USH1C de pérdida de función, que crea un críptico 5' del sitio de empalme en el exón 3, es único para los Acadianos. Los ratones genéticamente modificados para sufrir este tipo de síndrome sobrevivieron al huracán Katrina en Nueva Orleans y ahora sirven como modelo para el desarrollo de terapia en esta condición.
Los oligonucleótidos antisentido son cortos, químicamente modifican los ácidos nucleicos sintéticos que se unen al ARN por medio de la hibridación de pares de bases. Estos oligonucleótidos modulan la regulación postranscripcional, ya sea al silenciar los genes o al alterar el metabolismo del ARN. Se están desarrollando para tratar una variedad de enfermedades, incluyendo patologías neurodegenerativas, y algunos se encuentran en evaluación mediante ensayos clínicos. Debido a que la mutación USH1C Acadiana conduce a un defecto de empalme, el grupo de Jennifer J. Lentz diseñó un oligonucleótido antisentido que pretendía restaurar el correcto empalme.
El enfoque adoptado por los investigadores era engañosamente simple (figura 1) Utilizando pantallas in vitro, probaron decenas de oligonucleótidos antisentido por su capacidad de bloquear el sitio de empalme críptico y con ello la unión aberrante del ARN mensajero USH1C. Una vez que los oligonucleótidos eran capaces obstruir la unión críptica, se promovía el empalme correcto y el aumento de harmonina en las líneas celulares mutantes, las que estaban listas para poner a prueba el enfoque. El oligonucleótido antisentido "ganador" fue ASO-29, con la mayor capacidad de corrección y aumento de la expresión de harmonina. Los ratones mutantes fueron inyectados por vía intraperitoneal con 0,75 mg de ASO-29.
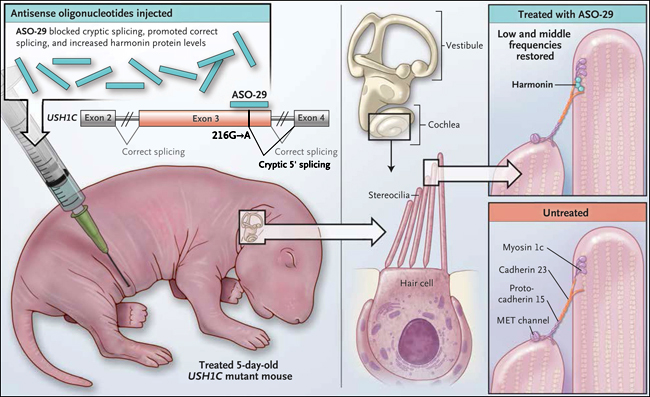
Figura 1: comportamiento y recuperación de la pérdida de audición en un modelo de ratón para el síndrome de Usher tipo 1
La mutación USH1C 216g→A introducida en ratones crea una sitio críptico de empalme 5' que es una réplica de la mutación USH1C Acadiana encontrada en seres humanos, la que se sabe causa pérdida auditiva, disfunción del equilibrio y ceguera. Los autores de esta investigación informan que la inyección intraperitoneal del oligonucleótido antisentido ASO-29 en estos ratones a los pocos días después del nacimiento conduce a la corrección del defecto de empalme y a la restauración funcional de la harmonina en las uniones de punta de los estereocilios, que contienen la miosina 1c, la cadherina 23, la protocadherina y los canales de transducción electro-mecánicos (MET). Los umbrales de audición de los animales tratados en 8 y 16 kHz fueron similares a los de ratones no mutantes, efecto que se mantuvo durante 6 meses. Las células basales de la cóclea se siguieron dañadas, y no hubo desarrollo de audición de alta frecuencia.
Los resultados fueron sorprendentes: audición normal y desarrollo del equilibrio en los animales mutantes tratados, los que también expresaban niveles normales de harmonina. Estos ratones también tenían más células pilosas y grupos celulares bien ordenados en comparación a los mutantes no tratados. La audición en frecuencias media y baja, en base a la respuesta auditiva del tronco cerebral, fue la misma que en no mutantes. Sin embargo, no se pudo restaurar la audición a altas frecuencias. Se observó una clara correlación entre el déficit de audición residual y el número estereocilios anormales en la base cóclear, un hallazgo con consecuencias desfavorables para la corrección de la pérdida auditiva de alta frecuencia asociada al envejecimiento y exposición al ruido. El momento de la aplicación de los oligonucleótidos fue crucial: de 3 a 5 días después del nacimiento fue más eficaz, a pesar que la función vestibular se recuperó cuando los ratones eran tratados 13 días después del nacimiento. Los resultados fueron duraderos: a los 6 meses de edad, la audición y la función vestibular se mantuvieron intactas.
¿Este enfoque es aplicable a otras formas de pérdida de audición? La discapacidad auditiva hereditaria es muy heterogénea. La terapia con oligonucleótido utilizada estaba dirigida a los defectos de empalme, responsables de sólo algunas formas de pérdida auditiva de carácter genético. ¿Cómo podría tal estrategia aplicarse en seres humanos? Dado que los recién nacidos humanos normales, a diferencia de los ratones, son capaces de escuchar al nacer, la ventana terapéutica para la terapia con oligonucleótidos podría darse durante la gestación, un período más complejo que podría complicar el proceso. Sin embargo, existe una gran cantidad de tiempo respecto al uso de oligonucleótidos para el tratamiento de la ceguera de aparición tardía en el modelo USH1C. La progresión relativamente lenta de la pérdida de visión en los seres humanos puede hacer que la prevención sea más susceptible de intervención.
Fuente bibliográfica
Rescue from Hearing Loss in Usher's Syndrome
Karen B. Avraham, Ph.D.
Department of Human Molecular Genetics and Biochemistry, Sackler Faculty of Medicine and Sagol School of Neuroscience, Tel Aviv University, Tel Aviv, Israel.
N Engl J Med 2013; 369:1758-1760