Edición de bases en la progeria
El síndrome de progeria de Hutchinson-Gilford es una enfermedad autosómica dominante causada por una mutación puntual en LMNA, que codifica las proteínas laminares A y C. Experimentos recientes han demostrado que la duración de la vida en un modelo de ratón de la enfermedad se prolongó mediante la edición de bases del ADN.
¿Cuánto tiempo más? Es una pregunta demasiado familiar para los padres. También se la hacen los científicos cuando intentan encontrar un tratamiento realmente eficaz para los pacientes con el síndrome de progeria de Hutchinson-Gilford (HGPS, por su sigla en inglés) y otras enfermedades genéticas. Koblan y colaboradores describieron recientemente una espectacular prolongación de la vida de un modelo de ratón de HGPS, un hallazgo que sugiere que la respuesta a esta pregunta podría ser "ya no falta mucho ".
El HGPS un trastorno prematuro autosómico dominante, es muy raro. En más del 90% de los niños que padecen esta enfermedad, la mutación etiológica es una sustitución de una citosina (C) por una timina (T) en la posición c.1824 de LMNA, que codifica la lámina A. Los síntomas se manifiestan durante los primeros años de vida e incluyen un grave fallo de crecimiento y piel esclerótica. A medida que la enfermedad progresa, se desarrolla aterosclerosis, contracturas articulares y displasia esquelética, así como pérdida de cabello, grasa subcutánea y células del músculo liso vascular. Los pacientes suelen morir prematuramente en la adolescencia como consecuencia de un accidente cerebrovascular o un infarto de miocardio. Aunque se han descrito estrategias de tratamiento en modelos preclínicos, se han probado algunas en ensayos clínicos y una ha sido aprobada recientemente por la Administración de Alimentos y Medicamentos, no existe cura.
Koblan y colaboradores describieron el uso de una herramienta de ADN en el tratamiento de esta enfermedad. Este enfoque genético implica el uso de CRISPR-Cas9 (repeticiones palindrómicas cortas agrupadas asociadas a la endonucleasa Cas9), que se descubrió originalmente como un sistema de defensa para la protección de las bacterias contra ADN extraño. El complejo CRISPR-Cas9 está formado por un ARN guía único (sgRNA) que dirige y ata el complejo a una secuencia específica de ADN. La enzima Cas9 corta el ADN objetivo, produciendo roturas de doble cadena. El descubrimiento de este sistema, ahora ampliamente utilizado por los investigadores, ha sido reconocido recientemente con el Premio Nobel de Química, concedido a Jennifer Doudna y Emmanuelle Charpentier.
El editor basado en adenina es una proteína de fusión compuesta por una desoxiadenosina desaminasa modificada y una variante de Cas9. Este editor de bases, al igual que la enzima Cas9, forma un complejo con un sgRNA. Una vez que el sgRNA se ha unido a su secuencia objetivo, el editor de bases, dentro de una ventana de edición de 4 a 5 nucleótidos, puede convertir una base de adenina (A) en una base de guanina (G). Una vez realizada la mella en una sola hebra, los mecanismos de reparación celular incorporan el par de bases A-T modificado en un par de bases G-C. El potencial terapéutico de la edición de bases de adenina se ha demostrado previamente en un modelo de ratón de distrofia muscular de Duchenne.
En el HGPS, la mutación LMNA c.1824C→T interrumpe el patrón normal de empalme del ARN mensajero (ARNm) del LMNA, lo que da lugar a la producción de una proteína laminar A más corta; esta proteína mutante se denomina progerina en referencia a la enfermedad. La progerina, a su vez, interrumpe muchas de las funciones de la lámina A codificadas por el alelo no mutante. Las lamininas se localizan en el núcleo y están implicadas en la organización de la cromatina, la expresión de los genes y replicación del ADN.
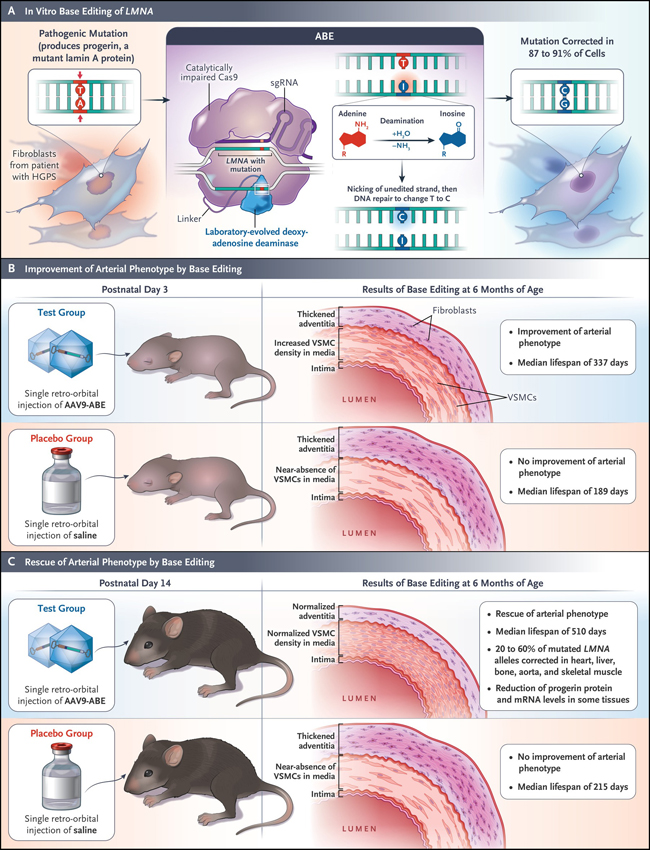
Figura 1: edición de bases en ADN en progeria.
Koblan y colaboradores informaron recientemente de la corrección in vitro e in vivo, mediante la edición de bases de adenina (ABE, por su sigla en inglés), de una mutación puntual en LMNA que causa la progeria. En los fibroblastos tratados de los pacientes, la mutación se corrigió en un 87 a 91% de las células. En un modelo de ratón de progeria con pérdida de células del músculo liso vascular (VSMC, por su sigla en inglés) de la aorta y fibrosis adventicia, el tratamiento con virus adeno-asociado (AAV) que codifica el ABE dio como resultado la preservación de las VSMC y fibrosis adventicia y una vida 2,4 veces mayor que la de los ratones inyectados con solución salina. AAV9-ABE indica el vector AAV de serotipo 9 que codifica ABE, HGPS al síndrome de progeria de Hutchinson-Gilford, mRNA al ARN mensajero y sgRNA a guía único de ARN.
Por medio de la edición de bases de adenina, Koblan y colaboradores corrigieron 87 a 91% de los loci mutantes en los fibroblastos de pacientes con HGPS in vitro, lo que dio lugar a una reducción en la expresión de progerina y una mejor estructura nuclear (Figura 1). Un análisis de 32 loci candidatos "off-target" en el ADN de estas células no mostró cambios sustanciales de nucleótidos. Además, el análisis de secuenciación de ARN de los fibroblastos HGPS editados mostraron un patrón de expresión génica similar al de los fibroblastos de un padre no afectado de un niño con HGPS.
Para analizar el potencial de su sistema in vivo, Koblan y colaboradores utilizaron un modelo de ratón que contenía dos copias de un transgén de LMNA humano mutado y que presentaba muchos síntomas de la enfermedad. Los genes que codifican el editor de bases y el sgRNA fueron entregados por dos construcciones, ambas utilizaban un vector de virus adeno-asociado (AAV) de serotipo 9, a través de una única inyección retro-orbital en el día postnatal 3 o 14 (figura 1). A los 6 meses en los ratones tratados en el día 14 postnatal, un total de 20 a 60% de alelos LMNA mutantes en la aorta, músculo esquelético, corazón, hígado y hueso fueron corregidos y los niveles de proteína progerina y su ARNm se redujeron en algunos de estos tejidos.
Los ratones tratados ("editados") tuvieron una vida más larga, más del doble que sus compañeros de camada inyectados con solución salina. Koblan y colaboradores han demostrado el potencial de la edición de genes en el tratamiento del HGPS. Sin embargo, es necesario realizar más pruebas preclínicas antes de que este enfoque pueda ser probado en pacientes. La dosis y el momento del tratamiento deben ser evaluados más a fondo, y deben investigarse métodos alternativos de entrega, así como enfoques que utilizan la expresión transitoria del editor de base y métodos de entrega no integrativos. Estas investigaciones también podrían centrarse en estrategias que prioricen tejidos o tipos específicos de células progenitoras que son importantes para la regeneración de tejidos.
En el estudio actual, se desarrollaron tumores de hígado en varios de los ratones tratados, y aunque la causa no está clara, una posible explicación es la integración mutagénica de un vector AAV en el ADN de los hepatocitos. No se han notificado tumores de hígado en seres humanos tratados con AAV; sin embargo, el tratamiento con AAV se ha asociado a tumores de hígado en ratones. No obstante, el uso de este sistema de administración de genes sigue planteando problemas de seguridad. Se ha detectado la edición no deseada del ADN (como inserciones o deleciones y la edición de vecinos) en bajas frecuencias en varios tejidos; no se ha investigado la edición y expresión del ARNm en los tejidos analizados. En conjunto, estas preocupaciones indican claramente que se necesitarían estudios adicionales antes de poder probar la edición experimental de bases en humanos. En otras palabras, aún no hemos llegado, pero estamos más cerca del destino.
Fuente bibliográfica
Base Editing in Progeria
Jin-Soo Kim, Ph.D., and Maria Eriksson, Ph.D.
Center for Genome Engineering, Institute for Basic Science, Daejon, South Korea (J.-S.K.); and the Department of Biosciences and Nutrition, Center for Innovative Medicine, Karolinska Institutet, Huddinge, Sweden (M.E.).
N Engl J Med 2021; 384:1364-1366