Antitrombóticos para el asma
En varios trastornos clínicos, se han observado interacciones entre la inflamación dependiente de la lesión de los tejidos y la formación de trombina, deposición de fibrina y la alteración de la fibrinólisis. La reciente evidencia generada a partir de un modelo de ratón para la hiperreactividad de las vías respiratorias alérgicas sugiere que la coagulación y la fibrinólisis pueden contribuir a la patogénesis del asma. Los mecanismos inflamatorios que conducen a la contracción del músculo liso bronquial y a la hiperreactividad de las vías respiratorias pueden estar asociados con la acumulación de fibrina extravascular, exudados de plasma y células inflamatorias que conducen al cierre de las vías respiratorias.
Asma y mecanismos de coagulación
El asma se caracteriza por la inflamación crónica de las vías respiratorias, contracción del músculo liso, la sobreproducción de moco y remodelación de la pared de los conductos respiratorios. En muchas de sus formas, hay una acumulación de eosinófilos, que predice una buena respuesta a los glucocorticoides inhalados. La inflamación eosinofílica es controlada por los linfocitos T helper tipo 2 (Th2) del sistema inmune adaptativo, que hacen que la interleucina-4, interleucina-5 y la interleucina-13 respondan a la presentación de un alérgeno por las células dendríticas en la vía aérea. Sin embargo, muchas características asmáticas también pueden ser controladas por células linfoides innatas tipo2, que no reconocen específicamente los alérgenos, pero sí producir interleucina-5 e interleucina-13 cuando son estimuladas por citoquinas producidas por las células y macrófagos epiteliales del pulmón.
Aunque las células inmunes controlan todas las características del asma, no está claro por qué y cómo reaccionan ante los alérgenos. La mayoría de los organismos que producen alérgenos, como las plantas de polinización, ácaros del polvo doméstico, cucarachas y hongos ambientales, no son intrínsecamente patógenos para el huésped. Sin embargo, algunas proteínas o lípidos de estos organismos activan los receptores de reconocimiento del sistema inmunitario de los mamíferos. Por ejemplo, el ácaro de polvo casero lleva al menos 20 alérgenos que pueden inducir respuestas de IgE en seres humanos. En ratones, el desarrollo de una condición parecida al asma provocada por los ácaros del polvo depende de la expresión de los receptores Toll-like 4 (TLR4) en las células epiteliales de las vías respiratorias. Aunque muchos alérgenos contienen lipopolisacáridos, que potencialmente pueden unirse y activar los TLR4, también es posible que TLR4 sean estimulados por señales endógenas de peligro que se liberan en la exposición a los alérgenos.
Algunos alérgenos tienen propiedades proteolíticas y pueden activar receptores estimulados por proteasas en células epiteliales de barrera y en células dendríticas, pero hasta la reciente publicación de un artículo de Valentine Ongeri Millien y colaboradores (Science 2013; 341:792-6), no se sabía de ningún vínculo entre los alérgenos proteasa y la señalización de TLR4. Estos investigadores encontraron que TLR4 tiene un papel crítico en la respuesta alérgica a las proteasas fúngicas, tales como las derivadas de las especies de Aspergillus del medio ambiente.
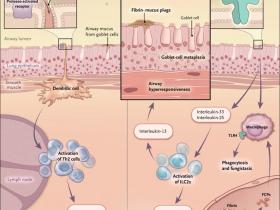
La colonización por Aspergillus se encuentra con frecuencia en pacientes con asma grave, y causa la aspergilosis alérgica broncopulmonar, un síndrome caracterizado por eosinófilos infiltrados pulmonares, alta producción de IgE y bronquiectasia central. El grupo de Valentine Ongeri Millien encontró que la inflamación de las vías respiratorias, hipersecreción de moco e hiperreactividad bronquial en respuesta a la inhalación de una proteasa de Aspergillus depende de TLR4 y sus moléculas adaptadoras (fig. 1). En comparación con ratones de control, los deficientes en TLR4, en la exposición a la proteasa de Aspergillus, mostraron un insignificante reclutamiento de células linfoides innatas tipo 2 (a pesar de un número normal de células Th2) y una reducción en las características asmáticas.
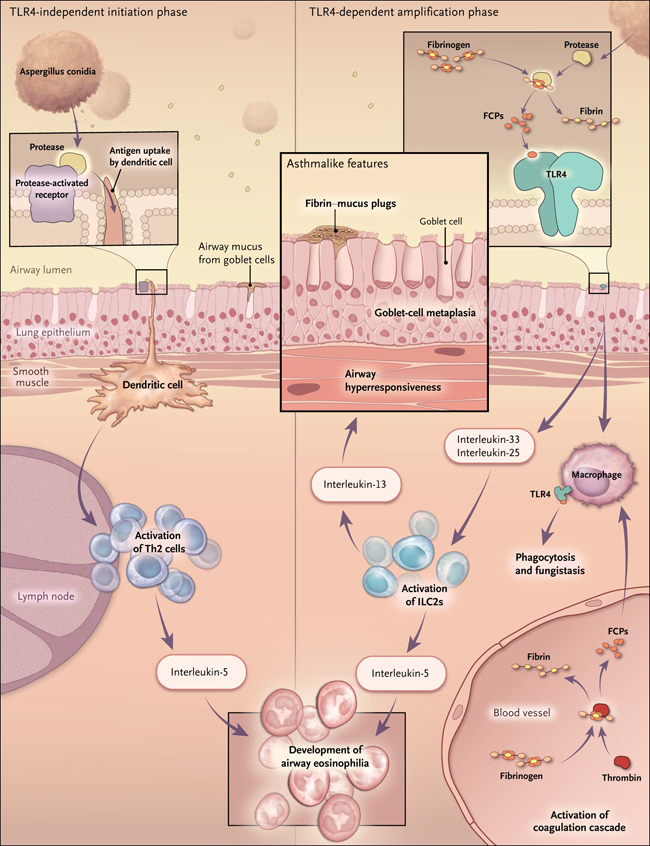
Figura 1: productos de escisión del fibrinógeno en el asma
Cuando se inhalan esporas de hongos o proteasas de Aspergillus, se desencadena la activación de células dendríticas generando inmunidad vía linfocitos T helper tipo 2 (Th2) en los ganglios linfáticos de drenaje. Los linfocitos Th2 luego regresan a los pulmones para causar eosinofilia leve y la producción de moco por las células caliciformes. Esta investigación determinó que las proteasas fúngicas posteriormente escinden el fibrinógeno en fibrina y productos de degradación del fibrinógeno (FCP). Los FCP activan el receptor tipo Toll 4 (TLR4) en las células epiteliales provocando la captación de células linfoides innatas tipo 2 (ILC2s), que aumentan aún más la eosinofilia y la producción de moco. La fibrina se une al moco para formar tapones que obstruyen los conductos respiratorios. Al mismo tiempo, la activación de TLR4 por FCP hace que los macrófagos inhiban el crecimiento de hongos. Este sistema en el que una molécula implica la coagulación por TLR4 es una reminiscencia de la vía Toll activada por Spätzle en moscas de la fruta.
Los investigadores también observaron que la activación de TLR4 en ratones tipo salvaje por una proteasa fúngica era indirecta y requería la presencia de un factor sérico. Al tratar de identificar el componente sérico responsable, los autores consideraron cómo se habían descubierto los receptores tipo toll en Drosófila. El ligando Toll pro-Spätzle es activado por una proteasa serina - un mecanismo que subyace también en la activación del coagulógeno, un factor de coagulación del cangrejo herradura, invertebrado que ha cambiado poco en los últimos 400 millones de años. También se encontró que, de manera similar, las proteasas fúngicas escinden el fibrinógeno, provocando la formación de coágulos de fibrinógeno y la liberación de productos de escisión que activan a TLR4. La estimulación de TLR4 por los productos de escisión del fibrinógeno posteriormente desencadena la inmunidad antifúngica y la fungistasis de Aspergillus conidia en los macrófagos alveolares, tal como sucede en Drosófila.
La trombina, el clásico activador de coagulación, también genera productos de escisión de partir de fibrinógeno, por lo tanto activa a los TLR4. El inhibidor de trombina hirudina es capaz de suprimir cambios asmáticos impulsados Aspergillus en ratones, y también controlar alteraciones provocadas por el antígeno de ovoalbúmina, que no tiene actividad proteasa. La estimulación alergénica en seres humanos y ratones generalmente se acompaña de la extravasación de plasma, agregación plaquetaria y la activación de la cascada de coagulación en el intersticio pulmonar y el compartimiento broncoalveolar, con elevaciones del factor tisular, de trombina y fibrinógeno, que explica cómo los productos de escisión del fibrinógeno pueden ser generados con la mayoría de los alérgenos.
Los investigadores encontraron que la activación de la cascada de coagulación por proteasas endógenas o derivadas de alérgenos es un factor importante en los cambios del asma en ratones por medio de la señalización de TLR4. Se requiere investigación adicional para evaluar si este hallazgo se puede traducir en aplicaciones clínicas. Es necesario definir con mayor precisión los productos de degradación del fibrinógeno antes que futuros fármacos puedan ser examinados. Queda por determinar si los productos de escisión del fibrinógeno están presentes en las vías respiratorias afectadas de las personas con asma. Aunque el grupo de Valentine Ongeri Millien fue capaz de eliminar las alteraciones por Aspergillus mediante el bloqueo de la producción de productos de escisión del fibrinógeno, es notable que los ensayos de inhalación de heparina (de bajo peso molecular) en seres humanos hayan producido diversos efectos. Dada la naturaleza heterogénea de la enfermedad, es concebible que un subconjunto de pacientes con asma puedan algún día beneficiarse de los medicamentos antitrombóticos.
Fuente bibliográfica
Asthma and Coagulation
Bart N. Lambrecht, M.D., Ph.D., and Hamida Hammad, Ph.D.
VIB Inflammation Research Center and Department of Respiratory Medicine, Ghent University, Ghent, Belgium.
DOI: 10.1056/NEJMcibr1311045