Agregados proteicos en la neurodegeneración
La agregación de proteínas y la disfunción del sistema ubiquitina-proteosoma son características distintivas de muchas enfermedades neurodegenerativas. Recientemente se ha estudiado el vínculo entre estos fenómenos empleando tomografía crioelectrónica para diseccionar la arquitectura molecular de los agregados de proteínas dentro de neuronas intactas con una alta resolución. Particularmente, los estudios se han centrado en los agregados poli-Gli-Ala (poli-GA) resultantes de la traducción aberrante de una repetición ampliada de secuencias GGGCC en el gen C9orf72, evento que es la causa genética más común de esclerosis lateral amiotrófica y demencia frontotemporal. La proximidad a las estructuras tipo lazo que forma poli-GA estabiliza una conformación transitoria de procesamiento de sustrato del proteosoma, lo que sugiere una degradación estancada. Por lo tanto, los agregados poli-GA pueden comprometer la proteostasis neuronal al impulsar la acumulación y el deterioro funcional de una gran fracción de los proteosomas celulares.
Bloqueo proteosomal
Las enfermedades neurodegenerativas a menudo se asocian con mutaciones genéticas que causan la repetición de secuencias cortas de nucleótidos. En los trastornos de la esclerosis lateral amiotrófica (ELA) y la demencia frontotemporal, tal expansión en una región no codificante de proteínas del gen C9orf72, conduce a productos de traducción aberrantes que contienen estiramientos repetitivos de residuos de glicina y aminoácidos de alanina. Estos productos "poli(GA)" forman agregados en las neuronas y se relacionan con la interrupción de un proceso celular clave en el que los complejos proteasomles degradan las proteínas. Sin embargo, la base bioquímica de esta alteración, y cómo podría promover la enfermedad, es poco entendida. Recientemente, Qiang Guo y colaboradores (DOI: 10.1016/j.cell.2017.12.030) trazaron con precisión las características organizativas y estructurales de los agregados poli(GA) y los complejos macromoleculares asociados en las neuronas usando una técnica llamada tomografía crioelectrónica 3D (cryo-ET), para proporcionar una visualización directa de cómo los proteasomas son interrumpidos por las proteínas de poli(GA).
La Cryo-ET 3D utiliza microscopía electrónica para ver secciones muy delgadas, congeladas pero hidratadas de una célula desde varios ángulos. Las imágenes resultantes se combinan para producir una imagen 3D llamada tomograma. Los autores usaron esta técnica para visualizar neuronas que habían sido genéticamente diseñadas para expresar un tracto poli(GA) que contenía 175 o 73 repeticiones. Los tractos se fusionaron con una proteína verde fluorescente que permitió determinar su posición de manera precisa mediante microscopía óptica correlativa. La proteína diseñada imita los tractos de poli(GA) que se producen a partir de la expansión de C9orf72, que tardan mucho tiempo en formarse in vivo. Los autores encontraron que las proteínas poli(GA) forman estructuras retorcidas altamente agrupadas y a menudo bifurcadas que son de espesor relativamente uniforme, pero de longitud y anchura variables, similares a las estructuras de poli(GA) previamente observadas por microscopía electrónica.
El valor del trabajo de los autores radica no sólo en su observación detallada de la estructura de los agregados de poli(GA) en las células, sino en su comparación con los agregados formados a través de una expansión genética diferente: una repetición de la glutamina (poly(Q)), que causa el trastorno neurodegenerativo de la enfermedad de Huntington, y que el mismo grupo analizó previamente. Esta comparación reveló diferencias estructurales que podrían explicar las diferencias en los mecanismos patogénicos entre las condiciones.
En primer lugar, los agregados formados en cada caso son estructuralmente distintos. Las proteínas poli(Q) forman estructuras fibrilares que muestran poca ramificación y están menos densamente empaquetadas que las cintas de poli(GA).
Segundo, cuando los autores usaron enfoques computacionales poderosos para buscar complejos macromoleculares conocidos en cada agregado, encontraron muchos proteasomas incorporados en agregados de poli(GA) (Fig. 1). De hecho, los datos bioquímicos sugieren que hasta un 50% de los complejos proteosómicos en la neurona se enredan altamente dentro de las cintas de poli(GA). La remoción de los proteosomas de su ubicación normal en las células a través de este mecanismo de secuestro podría explicar la reducción de la actividad proteosómica en células que albergan estos agregados. Los ribosomas, que median la síntesis de proteínas y son comparables en tamaño a los proteosomas, se excluyeron en gran medida de las cintas de poli(GA), lo que sugiere que los agregados de poli(GA) son reclutados o retenidos activamente por los proteosomas. Por el contrario, las fibrillas de poli(Q) no contenían proteosomas, sino que forman estrechos contactos con membranas de múltiples tipos de organelos. Esta interacción conduce a la deformación de las membranas alrededor de los organelos, como el RE. Tal deformación podría alterar las vías involucradas en la traducción, tráfico y degradación de proteínas.
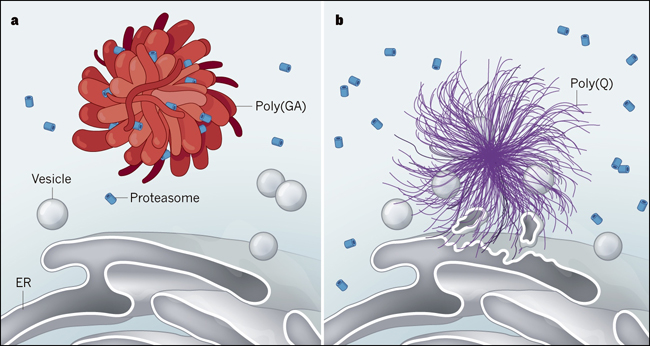
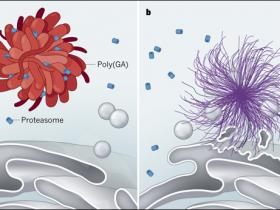
Figura 1. Mecanismos de contraste de la toxicidad agregada.
a, En algunos casos de la esclerosis lateral amiotrófica, cadenas largas de residuos de glicina y aminoácidos de alanina (denominados tractos poli(GA)) se agregan en las neuronas. Qiang Guo y colaboradores (DOI: 10.1016/j.cell.2017.12.030) muestran, a través de estructuras de alta resolución en las células, que los tractos poli(GA) forman agregados parecidos a cintas que capturan complejos proteicos llamados proteasomas, que normalmente procesan otras proteínas para su degradación. Tal captura causa el estancamiento del proteasoma, proporcionando una explicación para la toxicidad de este agregado. Los agregados de Poli(GA) no se unen a organelos asociados a membranas como las vesículas y el retículo endoplásmico (RE). b, Por el contrario, los tractos repetitivos del aminoácido glutamina (tractos poli(Q)), que están asociados con la enfermedad de Huntington, forman agregados similares a las fibrillas. Estos agregados deforman las membranas de las vesículas y del RE, sugiriendo que diferentes agregados causan neurodegeneración a través de diferentes mecanismos.
El proteasoma consiste en una estructura en forma de barril en cuyo centro se produce la ruptura de sustratos, y una o dos partículas reguladoras que cubren los extremos del barril, restringen el acceso al núcleo para que sólo puedan entrar proteínas marcadas con la molécula prodegradación ubiquitina. Se han observado partículas reguladoras en múltiples conformaciones lo que indica que los proteasomas progresan a través de un ciclo de reacción que involucra estados de sustrato,. Los autores también encontraron un gran aumento en la proporción de proteasomas de doble capa en comparación con las neuronas de control que no contenían productos de poli(GA). Casi un cuarto de los proteasomas dentro de los agregados adoptaron una conformación recientemente descrita como substrato comprometido pero estancado, en el cual el sustrato queda atrapado en el barril. Y esa proporción subió al 36% para los proteasomas más cercanos a las cintas de poli(GA).
¿Por qué ocurre este estancamiento? Las reconstrucciones tomográficas de los autores revelaron numerosas regiones de densidad de electrones localizadas entre una cinta de poli(GA) y el sitio donde la proteína RAD23 se une al proteasoma. RAD23 participa en el reclutamiento de proteínas marcadas con ubiquitina para el proteasoma y se sabe que está enriquecida en agregados de poli(GA). Por lo tanto, esta densidad de electrones podría indicar que la ubiquitina asociada con RAD23 que está unida a proteínas dentro del agregado. Actualmente, no está claro con qué proteína o proteínas se "asfixia" el proteasoma, aunque las posibilidades incluyen los mismos péptidos poli(GA) , los cuales probablemente inhiben directamente su actividad. Independientemente del mecanismo, parece probable que el agotamiento de la actividad proteasómica en la célula sería perjudicial para las vías de degradación de proteínas, contribuyendo así a la toxicidad celular.
Estos resultados plantean varias preguntas importantes. En primer lugar, la patogénesis en los casos de ELA impulsada por la expansión de C9orf72 se ha relacionado tanto con cambios mediados por la formación de poli(GA) como con cambios causados por la reducción de la producción de la proteína C9orf72, pero ¿cuáles son las contribuciones relativas de cada mecanismo? C9orf72 es parte de un complejo involucrado en la autofagia, un proceso por el cual el material celular, incluyendo las proteínas, es degradado y reciclado. Por lo tanto, es posible que la reducción de los niveles de C9orf72 conspire con la inhibición del proteasoma dependiente de poli(GA) para aumentar la toxicidad neuronal. Segundo, ¿la toxicidad es promovida por la captura de otras proteínas dentro de los agregados de poli(GA)? Uno de los candidatos es el receptor de carga de autofagia p62, que se sabe que se acumula en agregados de poli(GA). Tercero, varias organizaciones moleculares involucradas en el desmontaje de agregados no se acumulan en estructuras de poli(GA), pero las razones de esto no están claras.
Finalmente, aunque poli(GA) es la proteína repetitiva más abundante producida por la expansión de C9orf72, no es la única - la mutación también puede producir tractos de glicina-arginina (poly(GR)) y prolina-arginina (poly(PR)). ¿Cómo se comparan las estructuras de estos otros agregados con las de las proteínas poli(GA)? La mayoría de los datos sobre los agregados poli(GR) y poli(PR) indican que no acumulan proteasomas, lo que sugiere mecanismos alternativos de toxicidad. Un análisis más profundo con cryo-ET 3D, y el análisis de los productos naturales de la expansión de C9orf72 en lugar del producto de ingeniería utilizado en el presente estudio, podría aclarar las similitudes y diferencias entre los agregados que ocurren en los pacientes y las estructuras de agregados modelo estudiadas por Guo y sus colegas.
En resumen, el trabajo actual destaca la resolución sin precedentes de la crio-ET 3D para la visualización de procesos fundamentales dentro de las células. Además, sienta las bases para una comprensión mecanística más completa de las enfermedades neurodegenerativas asociadas los agregados proteicos.
Fuente bibliográfica
Protein aggregates caught stalling
Laura Pontano Vaites & Wade Harper
Department of Cell Biology, Harvard Medical School, Boston, Massachusetts 02115, USA.
DOI: 10.1038/d41586-018-03000-2