Implicaciones clínicas
Huntington: una molécula, varios caminos
INTRODUCCIÓN
La enfermedad de Huntington (EH) es una condición hereditaria caracterizada por movimientos corporales anormales, demencia y problemas psiquiátricos. Es un trastorno progresivo que ocasiona debilitamiento (degeneración) de las células nerviosas en el cerebro y fue descrita inicialmente en 1872 por George Huntington (1850-1916), un médico estadounidense. Afecta a ambos sexos por igual. También se le ha denominado Corea (del griego danza) por el movimiento característico de las personas afectadas y durante mucho tiempo igualmente ha sido conocida como “Baile de San Vito”.
El trastorno se hereda como un gen defectuoso único en el cromosoma 4 (4p16.3). Una parte del gen se repite en múltiples copias. Cuanto mayor sea el número, mayores serán las probabilidades de que se desarrollen los síntomas y que se presenten a una edad más temprana. Puede presentarse cada vez más temprano y de manera más severa en cada generación subsiguiente afectada, ya que el número de copias tiende a aumentar. El diagnóstico diferencial es amplio cuando el caso se presenta aislado. Su curso es progresivo y fatal.
Cada hijo de un padre que presente el trastorno tiene un 50% de posibilidad de heredar la enfermedad de Huntington. La sintomatología generalmente no aparece hasta la adultez, normalmente entre los 35 y 50 años; sin embargo, esto depende del número de copias encontradas en el gen, así que el trastorno puede aparecer en personas más jóvenes. En los niños, puede presentarse como la enfermedad de Parkinson con rigidez, movimientos lentos y temblor. Se presenta pérdida progresiva de la función mental que involucra cambios de personalidad y pérdida de las funciones cognoscitivas como el juicio y el lenguaje. También se desarrollan movimientos faciales y corporales anormales, entre ellos, rápidos movimientos espasmódicos.
No existe cura para la patología y no hay forma conocida de detener su progresión. El manejo se orienta a reducir su progresión y a maximizar la capacidad funcional tanto como sea posible. La medicación varía de acuerdo con los síntomas. Los bloqueadores de la dopamina, como haloperidol o fenotiacina, pueden reducir los comportamientos y movimientos anormales. También se ha usado reserpina y otros medicamentos con éxito variable. Drogas como la tetrabenazina y amantidina se utilizan para tratar de controlar los movimientos adicionales. Hay evidencias que sugieren que la co-enzima Q10 puede disminuir algo la evolución. Las patologías siquiátricas, la depresión y el suicidio son comunes durante la enfermedad de Huntington. El tratamiento sintomático para la demencia es similar al usado para cualquier síndrome cerebral orgánico. Inicialmente, la señalización para recordar cosas y otras ayudas puede mejorar la función de la memoria.
BASES MOLECULARES Y POSIBLES MECANISMOS PATOGÉNICOS
¿Por qué tiene importancia la enfermedad de Huntington? Sobre todo por dos razones. La primera, porque es una enfermedad de pronóstico terrible, hereditaria y cuya prevalencia no es tan baja, 10 pacientes por cada 100.000 habitantes, en muchos países occidentales más frecuente que otras enfermedades superpopulares como el SIDA. La segunda razón, es porque la patología es el prototipo de enfermedades por expansión inestable de tripletes de ADN, abriendo muchas líneas de trabajo sobre el papel que juegan determinadas proteínas y fragmentos de proteínas en la función celular cerebral.
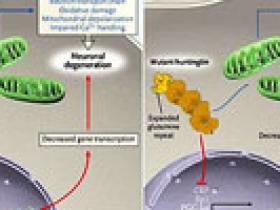
Cuando el gen causativo de la enfermedad de Huntington fue identificado en 1993, había gran expectación y esperazas que los mecanismos provocadores de la condición serían rápidamente identificados y que los respectivos tratamientos neuroprotectivos pronto aparecerían. Catorce años más tarde, es obvio que tal postulado era incorrecto. Los potenciales mecanismos patógenos han proliferado, y su importancia relativa es incierta. Ahora, tres estudios recientes han identificado una proteína y un mecanismo que unen dos de las principales hipótesis de la patogénesis: desregulación transcripcional y debilitación mitocondrial (figura 1).
La enfermedad de Huntington se genera por sucesivas repeticiones de trinucleótidos CAG en el exón 1 del gen que codifica la proteína huntingtina (HTT) de 348 kDa. Si el gen contiene más de 35 CAG, que codifican para un residuo de glutamina, la enfermedad de Huntington es muy probable que aparezca. La huntingtina mutante, en virtud de la cadena expandida de residuos de glutamina, ata ciertos factores de transcripción (o a las proteínas que interactúan con factores de transcripción), limitando la funcionalidad de las mismas poliglutaminas. Por ejemplo, se ha visto que la huntingtina interactúa con otra proteína llamada CBP (proteína que se une al factor de transcripción CREB). La CBP se encuentra involucrada en la supervivencia celular en neuronas normales. La huntingtina (que es una molécula grande) secuestra y desplaza a la CBP (que es pequeña), lo que produce que la CBP deje de actuar y se produzca toxicidad neuronal. A mayor número de repeticiones CAG (más grande es la molécula) mayor toxicidad. De una manera similar, la HTT alterada interfiere con la transcripción del gen Sp1. Así, la mutación puede modificar el complemento de proteínas que se sintetizan en una célula, un cambio que termina en el patrón neurodegenerativo característico de la EH.
Una hipótesis competente es que la neurodegenaración de la condición resulta de la debilitación mitocondrial. De hecho, existe mucha evidencia que demuestra las anormalidades bioquímicas, morfológicas y mitocondriales en los pacientes con esta enfermedad. Las actividades de los complejos mitocondriales del transporte de electrones II, III y IV se reducen en la enfermedad de Huntington, y la espectroscopia por resonancia magnética del cerebro muestra elevados niveles de lactato, consistente con la disfunción mitocondrial. Cómo se presenta la debilitación mitocondrial es incierto, pero un efecto directo de la huntingtina anormal en la mitocondria puede ser en parte la razón. ¿La transcripción alterada del gen podría ser también causa de la disfunción mitocondrial? Después de todo, la gran mayoría de las proteínas mitocondriales son codificadas por el genoma nuclear y se transportadas a los organelos, pero ¿qué regula su transcripción?
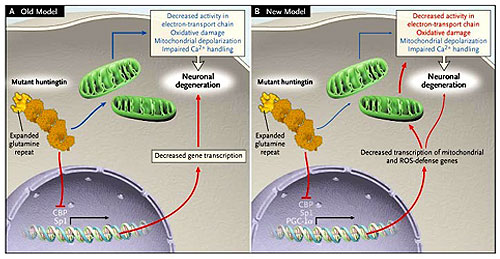
La enfermedad de Huntington se asocia a la desregulación transcripcional (debido a la interferencia en la transcripción mediada por Sp1 y la proteína de unión al elemento de respuesta de AMPc [CREB], indicada por las flechas rojas) y a la disfunción mitocondrial (indicada por flechas azules). Se creía que estas dos anormalidades se daban por efectos independientes de la huntingtina mutante (panel A). Sin embargo, recientes estudios apoyan un efecto interdependiente con actividad reducida del receptor activado de proliferación de peroxisoma (PGC-1-alfa), que regula la expresión de muchas proteínas mitocondriales nucleares y de las proteínas que proporcionan protección contra especies reactivas del oxígeno (ROS). Estas investigaciones demuestran que en parte la debilitación mitocondrial observada en la enfermedad de Huntington pueda estar causada por la desregulación transcricional (panel B).
El receptor activado de proliferación de peroxisoma (PGC-1-alfa) es un coactivator transcripcional que controla muchos procesos metabólicos, incluyendo la biogénesis mitocondrial, la fosforilación oxidativa y la termogénesis adaptativa (la respuesta del cuerpo a temperaturas frías). Este regula la expresión de una gran cantidad de genes, incluyendo a las subunidades codificadas en el genoma nuclear de cada uno de los complejos de la cadena transportadora de electrones y a varios genes que proporcionan protección contra el efecto de las especies reactivas del oxigeno (ROS). Como tal, el PGC-1-alfa parece situarse como puente entre la desregulación transcripcional y la disfunción mitocondrial, según lo sugerido recientemente.
Investigaciones adicionales han reportado que la HTT mutante reprime la expresión del gen que codifica al PGC-1-alfa uniéndose a su promotor e interfiriendo con la transcripción CREB-dependiente (proteína de unión al elemento de respuesta de AMPc). Weydt y colegas han reportado una menor expresión estriatal de PGC-1 alfa, una observación que es concordante con otros ensayos. Por otra parte, se ha demostrado que las células que no expresan PGC-1-alfa tienen un deteriorado sistema de defensa contra ROS al tener alterada la función antioxidante. La evidencia quizás más fuerte y más directa es que la menor actividad de PGC-1-alfa resulta en una neurodegenración es que su mayor expresión mejora la atrofia de las neuronas estriatales que normalmente ocurre en ratones transgénicos con la enfermedad de Huntington.
Por lo tanto, la idea básica es que en pacientes con EH, la transcripción del gen que regula PGC-1-alfa es defectuosa. Consecuentemente, hay expresión reducida de los genes mitocondriales y antioxidantes regulados por PGC-1-alfa. De esta manera, el coactivador transcripcional proporciona un entendimiento creíble entre mecanismos previamente sin relación: desregulación transcripcional y debilitación mitocondrial. Se ha propuesto que la manipulación de PGC-1-alfa pueda ser terapéutica en otras enfermedades neurodegenrativas en las cuales la presión oxidativa sea patógena. Se ha observado que en ratones carentes del gen PGC-1-alfa tienen aumentada la sensibilidad a la neurotoxina 1-metil-4-fenil-1,2,3,6-tetrahidropiridina (MPTP) inductora del parkinsonismo e, inversamente, que las células nerviosas que expresa de forma exógena a PGC-1-alfa son comparativamente resistentes al estrés oxidativo.
Los estudios subrayan el papel de PGC-1-alfa en la neurodegeneración. Ellos realzan la posibilidad que potenciar la expresión o la función de PGC-1-alfa podría tener un efecto terapéutico en la enfermedad de Huntington y otros desórdenes neurodegenarativos. Lamentablemente, hay que moderar el entusiasmo sobre este acercamiento ya que se ha observado que la sobreexpresión de PGC-1-alfa en el corazón causa incontrolada proliferación mitocondrial, desorganización de la estructura sarcomérica y cardiomiopatía dilatada. Estudios adicionales sobre PGC-1-alfa seguramente conducirán a nuevas estrategias de tratamiento que retrasen o retarden el impacto de las enfermedades neurodegenerativas. El hecho de que la interrupción de la producción de la HTT mutante en modelos transgénicos condicionales conduzca a una reversión del fenotipo patológico ofrece nuevas esperanzas sobre la posible intervención terapéutica en la EH.
Fuente bibliográfica
Huntington's disease - making connections
J. Timothy Greenamyre, M.D., Ph.D.
Pittsburgh Institute for Neurodegenerative Diseases and the Department of Neurology, University of Pittsburgh, USA.
N Engl J Med. 2007 Feb 1;356(5):518-20