Senescencia celular oncogénica
Alguna vez un crecimiento canceroso, ahora un lunar
INTRODUCCIÓN
Una nueva y sorprendente conclusión de la investigación biomédica: resulta que cada lunar sobre nuestra piel es un tumor de células pigmentadas que iniciaron un camino hacia cáncer y luego se detuvieron. En resumen, los lunares serían cánceres incipientes que por alguna razón y en algún momento del proceso neoplásico se truncaron.
Esto es una visión que ha sorprendido a muchos científicos. Los lunares son una especie de híbridos extraños en el espectro oncológico. Son pequeños tumores benignos, principalmente inofensivos, con células que no pueden extenderse por el cuerpo como un tumor canceroso y matar a una persona. Se inician con mecanismos idénticos a los descritos para los tumores malignos, con un gen que se transforma y deja a las células proliferar desordenadamente. Para el doctor Wolter J. Mooi, patólogo de la Universidad Vrije en Holanda, los lunares son verdaderos tumores, de hecho, si este tipo de células no dejaran de crecer, no sólo tendríamos lunares muy grandes, sino que muchos hubiesen progresado a melanomas totalmente malignos.
Algunos científicos sospechan que el mismo tipo de fenómeno también pasa dentro del cuerpo, con tumores diminutos que van camino al cáncer y luego se detienen para siempre. Ellos por lo general pasan inadvertidos porque son muy pequeños, nadie los busca, y no tienen ningún tipo de pigmento reconocido. Esto es una idea que sorprende: una célula sufre una mutación al azar en un gen que la dirige hacia un proceso cancerígeno, por consiguiente, la célula comienza a dividirse, entonces, la transformación se detiene y el tumor nunca podrá replicarse nuevamente. Pero por sobre todo, este singular proceso puede significar un poderoso mecanismo de defensa para hacer frente a los diferentes cánceres. Si bien no sería el único modo de parar un proceso neoplásico, se muestra sumamente eficaz dado la cantidad de lunares que muchas personas tienen. De hecho, es tan eficiente que la mayoría de los melanomas raramente se originan de los lunares, por el contrario, suelen provenir de otras células de la piel.
Pocos habrían predicho que una célula podría comenzar el complejo y largo camino del cáncer para luego pararse, y cuando esto de sugirió por primera vez, el tema se observó con mucho escepticismo. Afortunadamente, esto ha cambiado radicalmente, las últimas investigaciones han demostrado que el fenómeno explicaría también la existencia de pequeños tumores similares a los lunares en próstatas humanas. Ahora, la comunidad de científicos y médicos quieren desarrollar tratamientos para el cáncer que imiten el proceso.
En el presente artículo, W. J. Mooi y D. S. Peeper explican el fenómeno que se da en los lunares. Las células tienen un gen del cáncer, el BRAF, que las hace dividirse. El BRAF entonces se relaciona con otro gen, el p16, que a su vez las hace dejar de dividirse.
EL ALTO EN LA RUTA AL CÁNCER
En muchos tejidos hay numerosos pequeños islotes de lesiones neoplásicas que raramente se convierten en cánceres evidentes. En ellas se han encontrado procesos de clonación y mutación oncogénica. Estas lesiones incluyen a los nevos melanocíticos u otros tumores benignos los que histológicamente son solo marginalmente anormales. Llegados a cierto tamaño dejan de crecer y tampoco se hacen más agresivos durante años o décadas.
El progreso de un fenotipo maligno requiere múltiples mutaciones oncogénicas lo que no ocurre frecuentemente. Además, las lesiones sin capacidad para producir un estroma vascular raramente crecen más allá de algunos milímetros cúbicos. Por otra parte, la apoptosis puede ser producida por el proceso de proliferación celular oncogénico directamente ó a través de la activación de células “asesinas” u otras células inmunes. Un fenómeno relacionado, es la ausencia de figuras mitóticas en muchos tumores benignos, como el nevos melanocítico ó lipomas. Los tumores benignos son proliferaciones clonales pero en su interior albergan algunas mutaciones oncogénicas. Estas en algún momento deben cesar en su evocación de señales proliferativas. En tumores malignos, muchas de estas células salen del ciclo proliferativo. Este fenómeno, que no es bien comprendido, tiene un efecto obvio sobre la cinética del cáncer (figura 1).
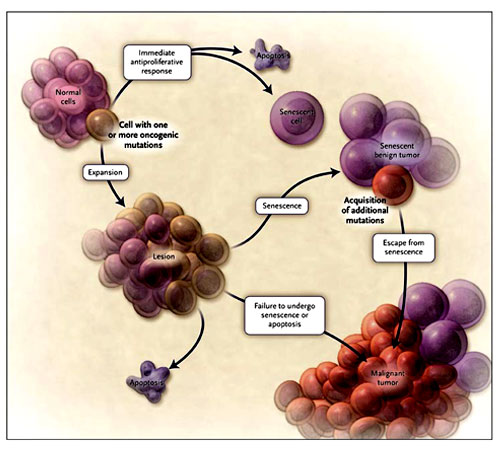
Cuando una célula adquiere una mutación oncogénica, ésta puede seguir tres tipos de respuesta. Una antiproliferativa, que provoca la muerte programada (apoptosis) o su senectud. Alternativamente, en ausencia de una respuesta inmediata, la proliferación por la mutación puede producir una lesión. En esta etapa, pueden activarse la apoptosis y la senectud, dando por resultado la muerte celular o el envejecimiento. En ausencia de mecanismos de defensa apropiados, el continuo crecimiento sumado a eventos genéticos puede conducir a una lesión maligna. Si las células experimentan una respuesta senil, pueden mantenerse por décadas, generando los nevos melanocíticos, pero también pueden escaparse de su estado senescente, aunque infrecuentemente, y transformarse en un tumor maligno.
La investigación sobre los efectos de las señales mitogénicas manejadas por los oncogenes pueden tener aplicaciones diagnósticas y pronósticas y pueden llevarnos a entender los mecanismos que detienen el crecimiento neoplásico al mismo tiempo que pueden señalarnos nuevas estrategias para la prevención y tratamiento del cáncer. Últimamente se ha descubierto un intrigante mecanismo de cese de crecimiento de tumores premalignos o benignos, en que, en ciertas circunstancias las señales oncogénicas pueden elegir paradójicamente un cese de crecimiento al cambiar el fenotipo celular a otro llamado “senescente”. Los últimos avances en este campo indican que la senescencia oncógeno-inducida es un mecanismo in vivo que contribuye a la protección contra el cáncer.
Existen ciclos definidos y específicos para la integridad del genoma y la supervivencia celular. La diferenciación celular y la apoptosis están sujetas a estrictas regulaciones; cuando estas se rompen estamos entrando al camino del cáncer, con signos aumentados de crecimiento celular, señales antiproliferativas alteradas y deficientes señales apoptósicas. Se ha observado que in-vitro ciertas células dejan de proliferar. Este fenómeno se conoce como “límite Hayflick”. Las células se hacen largas delgadas y vacuolares y activan concomitantemente con su condición de senescencia el ácido beta-galactosidasa, el que se puede mostrar con una reacción citoquímica que produce precipitaciones de color azul (figura 2).
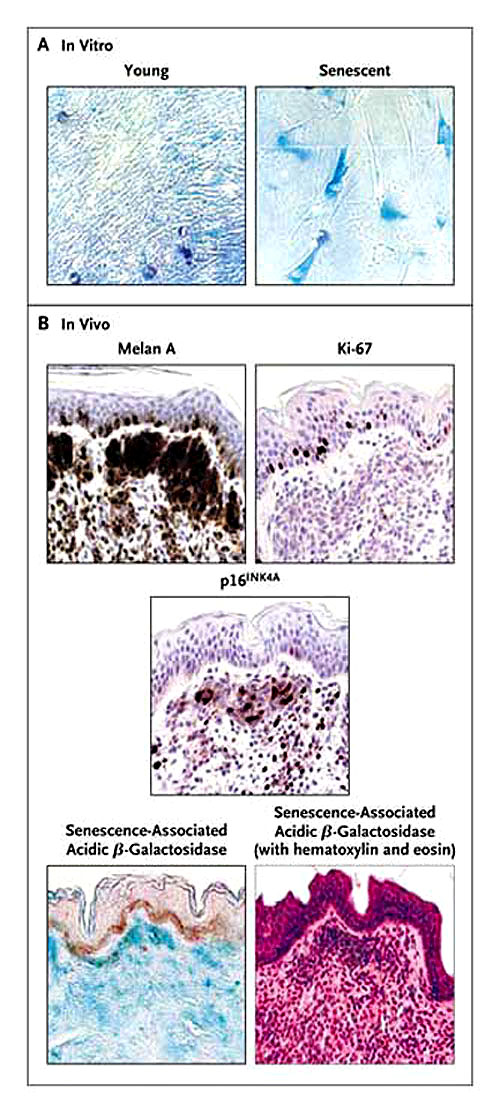
El panel A muestra un cultivo de fibroblastos humanos in vitro en estado joven y senescente, este último asociado a ácido beta galactosidasa que produce una precipitación azul. En el tejido joven, éstas son escasas y las fibras delgadas. En el estado senescente, las fibras son largas y delgadas, multinucleadas y con amplias precipitaciones azules asociadas al aumento del ácido beta galactosidasa. El panel B señala cortes histológicos de un lunar humano (todas las imágenes son de la misma lesión). El melano A de color marrón identifica las células del nevo. El marcador de proliferación Ki-67 (marrón) no se expresa en las células intradérmicas del nevo que son positivas para melano A. Algunos queratinocitos epidérmicos basales son positivos. El anticuerpo p16INK4A detecta p16INK4A (marrón), con resultados positivos en algunas células, pero no en otras. Las áreas azules en la dermis representan grupos celulares del nevo; esto es especialmente evidente en la tinción con hematoxilina y eosina; y en la senescencia asociada al ácido beta-galactosidasa.
El límite Hayflick resulta del hecho que después de cada replicación del ADN, los telómeros se hacen más cortos hasta llegar a una longitud crítica en que se gatilla un respuesta de daño del ADN, lo que está asociado con la vejez celular. Este fenómeno puede ser revertido por la enzima telomerasa, la que puede aumentar el número de divisiones celulares. El acortamiento de los telómeros puede ser un medio natural de evitar el crecimiento del cáncer ya que estas células pueden haber llegado a su máximo permitido de divisiones, haciendo del cáncer una enfermedad autolimitada, salvo por la inapropiada activación de la telomerasa u otro mecanismo similar que alargue los telómeros. Por otro lado, las senescencia celular incluye muchos más factores que la disfunción de los telómeros. Esta puede ser causada por estrés celular debido a daño del ADN, factores del cultivo celular o por activación oncogénica.
En 1997 Serrano y colaboradores descubrieron que mutaciones del gen RAS en fibroblastos jóvenes producían varios tipos de senescencia celular e incluso detención de los ciclos celulares. Este tipo de senescencia es irreversible, distinto del caso en que hay disminución del factor de crecimiento que acompaña la diferenciación celular que es de por sí fisiológica. En los cultivos celulares la “senescencia prematura” es activada por un conjunto de genes tumor- supresores que están inactivados en el cáncer humano. Así se llega al p16 que bloquea la entrada de la célula de la fase S del ciclo celular. La inactivación de estos genes supresores, es sine quanon en la tumorogénesis humana.
Cabe la duda que lo que sucede in vitro no sea igual a lo que pasa in vivo. Por eso se está estudiando la senescencia celular en ratas transgénicas. Pandolfi encontró que la senescencia in vivo no solo se puede producir por activación oncogénica sino también por inactivación de los supresores tumorales. Esta supresión se produce a través del p53, p16, p19 y otros.
La elevación de p16 no parece ser un prerrequisito para el envejecimiento de las células del mevi, ya que ésta también ocurre cuando el p16 está bajo o no existe. La senescencia inducida por niveles fisiológicos BRAF, sin relación con el p16 se puede reproducir in vitro al menos en fibroblastos benignos. En el Síndrome Nevos de Leiden Displástico, estos son más grandes y numerosos lo que se asocia a falta de p16, por lo que se supone que este limita la proliferación del los nevos melanocíticos, debiendo contribuir con lo otros factores supresores a la respuesta proliferativa.
Se ha observado en forma colectiva que en los inicios de una tumorogénesis, o sea, antes que la inestabilidad genómica emerja y se transforme en múltiples aberraciones genéticas la células de los mamíferos responden con señales que producen estrés en la replicación del ADN o disfunción telomérica activando redes protectoras que disminuyan la proliferación del cáncer (figura 3).
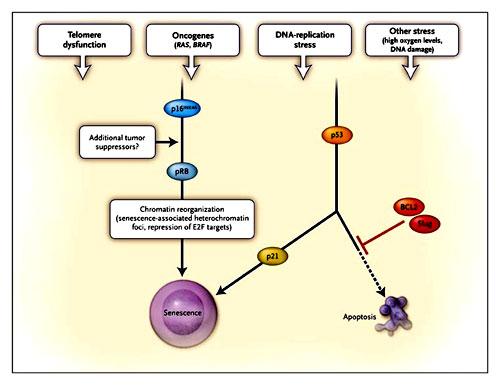
Hay varios tipos de estrés que gatillan el proceso supresor. Las células elevan los niveles de p16. De este modo la proteína retino blastoma (pRB) se acumula en estado activo. De esta forma se activa una cadena supresora que impide la replicación del ADN. El segundo paso tumor supresor activado por el estrés es el p53 que en su secuencia incluye genes antiproliferativos como el p21 y varios genes pro apotósicos. El mecanismo determina la producción de p53. Los niveles abundantes de BCL2 y “slug” encontrados en los melanocitos promueven el equilibrio hacia la senescencia.
¿Cuál podría ser la razón de la aparición de la senescencia oncogénica en la evolución cuando existe la apoptosis oncogénica? Podría ser el mantener melanocitos que están permanentemente expuestos al estrés por la luz ultravioleta, que son de larga vida y difíciles de reemplazar adecuadamente. Estos melanocitos producen abundante BCL2, una proteína anti–apoptósica que les permite sobrevivir y que en su estado de senescencia evitan el peligro oncogénico. Adicionalmente, estos melanocitos con BCL2 expresan “slug” que los defiende de la apoptosis dependiente del p53. Sin embargo, la respuesta senescente promueve la sobrevida protegiéndolos del cáncer, pero podría por otra parte interferir con la sobrevida general a largo plazo por la acumulación de células envejecidas.
¿Las mutaciones de líneas germinales o polimorfismo que influencia la senescencia oncogénico-inducida juegan algún rol en el cáncer? Desde un punto de vista terapéutico estudios en el cáncer mamario sugieren que la transformación maligna puede no impedir completamente la capacidad de producir una respuesta senescente después de una terapia citotóxica y por ende la apoptosis disminuida contribuya a la sobrevida del tumor durante el tratamiento o a su recidiva. Las implicaciones clínicas de estos hallazgos serán intensos focos de investigación en los años venideros y tal vez nos permitan comprender mejor la cinética y los riesgos de tratamiento del cáncer.
Fuente bibliográfica
Oncogene-induced cell senescence--halting on the road to cancer
Mooi WJ, Peeper DS.
Department of Pathology, Vrije University Medical Center, Amsterdam, The Netherlands
N Engl J Med. 2006 Sep 7; 355(10):1037-46