Terapia molecular para la atrofia muscular espinal
La atrofia muscular espinal (AME) comprende un grupo de trastornos autosómicos recesivos que se caracterizan por una debilidad progresiva de las neuronas motoras inferiores.
En la década de 1980, Werdnig y Hoffman describieron un trastorno de debilidad muscular progresiva que se iniciaba en la infancia, provocando una muerte temprana. En términos patológicos, la enfermedad se caracteriza por la pérdida de células del asta anterior. El efecto central de la degeneración de las neuronas motoras inferiores fue confirmado en posteriores estudios patológicos que demostraban la pérdida de células del asta anterior de la médula espinal y los núcleos de los pares craneales.
Desde entonces, varios tipos de atrofias musculares espinales se han descrito en base a la aparición de las características clínicas que los acompaña. Las formas más comunes son la aguda infantil (SMA tipo I, o enfermedad de Werdnig-Hoffman), infantil crónica (atrofia muscular espinal tipo II), juvenil crónica (SMA tipo III o enfermedad de Kugelberg-Welander) y la aparición en el adulto (atrofia muscular espinal tipo IV). Los defectos genéticos asociados con los tipos de SMA I-III se localizan en el cromosoma 5q11.2-13.3.
Terapia antisentido para la AME
La principal característica de la atrofia muscular espinal recesiva autosómica es la muerte de las neuronas motoras en la zona posterior, lo que causa parálisis generalizada y, en su forma más grave y frecuente (tipo 1), una grave insuficiencia respiratoria. Consecuencia directa de este resultado, es que la corrección de la deficiencia central en la neurona motora podría tratar o incluso prevenir la enfermedad. Por lo tanto, la neurona motora sería el blanco natural para el manejo eficaz de la atrofia muscular espinal.
Ahora, un reciente informe de Yimin Hua, Krainer y colaboradores han puesto en duda la simplicidad de este modelo, pero aportan información importante para el uso de oligonucleótidos antisentido (ASO, por sus siglas en inglés) en el tratamientos de enfermedades genéticas hereditarias. La mayoría de los casos de atrofia muscular espinal son causados por la pérdida de SMN1. Dada la centralidad de la proteína llamada factor de supervivencia de las neuronas motoras (SMN) en la biología del ARN, incluyendo una función crítica de unión, su pérdida es letal en todas las especies salvo en humanos, quienes, únicamente, tienen (en variado número de copias) un homólogo parálogo: SMN2. Aunque SMN2 codifica la proteína SMN, una transición C → T en el exón 7 produce la escisión de este exón, pero no en todos, de los ARN mensajeros (ARNm) de SMN2, resultando la pérdida neta de aminoácidos 8 carboxi-terminal (SMN contiene normalmente 294 aminoácidos), evitando así proteínas truncadas en la oligomerización (fig. 1). La no oligomerización genera SMN truncada, que es inestable y se degrada rápidamente. La SMN2 produce una pequeña cantidad de ARNm SMN2 de longitud completa, generando un mayor número de copias de SMN2 y por lo tanto, la condición más suave (en general) de atrofia muscular espinal observada en los pacientes. Provocar que SMN2 genere más copias completas del ARNm de SMN como medio de tratamiento de la atrofia muscular espinal, es el objetivo de varios laboratorios.
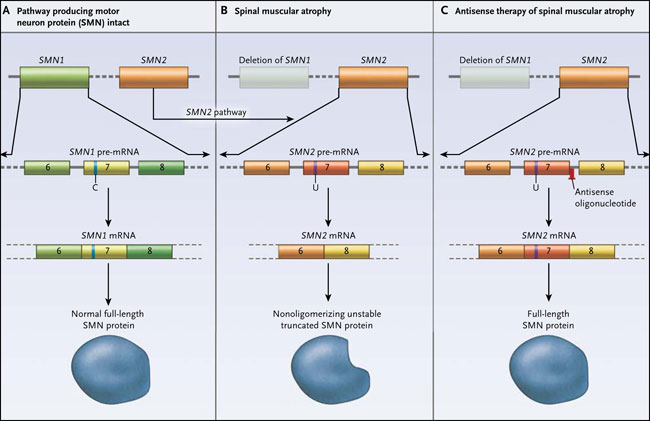
Los genes SMN1 y su parálogo SMN2, codifican a la proteína de supervivencia de las neuronas motoras. La presencia de citosina (C) o uracilo (U) en un sitio crítico del exón 7 resulta en la inclusión (panel A) o exclusión (panel B) del exón 7 en el ARNm de SMN. La proteína SMN2 codificada por el ARNm al que le falta el exón 7 se trunca y no se oligomeriza, lo cual la hace inestable. El enmascaramiento de un sitio de empalme intrónico en el exón 7 con un oligonucleótido antisentido reprime la escisión del exón 7, aumentando el ARNm de SMN de longitud completa y generando proteínas (panel C). La SMN es la proteína de supervivencia de las neuronas motoras.
Adrian R. Krainer y colegas, anteriormente habían demostrado que un derivado de ASO dirigido a una región específica de una variante patógenica de SMN2 podía enmascarar una secuencia de empalme intrónico, resultando in vitro e in vivo la reconstitución de un ARNm "normal" de SMN con longitud completa, y por lo tanto, la proteína, con niveles fisiológicos casi normales (fig. 2). El ASO fue de suficiente longitud para disminuir los efectos, pero no tanto como para restringir la biodistribución. Más recientemente, el grupo de A. Krainer demostró que una sola inyección intracerebroventricular de este ASO el primer día de vida en ratones con atrofia muscular espinal severa permitía duplicar su vida útil de 10 días. De forma más impresionante, se observó la extensión de la vida en al menos 100 días después de dos dosis más altas de ASO administradas por vía subcutánea o por vía intraperitoneal en los 3 primeros días de vida. Ahora, en el mejor de los casos, la corrección del empalme de SMN2 en la médula espinal es modesta. La superioridad de la dosis sistémica sobre la intracerebroventricular para un trastorno en el cual las neuronas motoras deberían ser el principal y único sitio, posiblemente, no fue claramente anticipada y pone en tela de juicio la primacía de los niveles de SMN sobre la neurona motora en la fisiopatología de la atrofia muscular espinal.
Estos resultados están en contradicción con otros estudios que han demostrado un rescate impresionante en los modelos de ratón para la atrofia muscular espinal por medio de la genética o por reposición de SMN mediado por ASO en el sistema nervioso central, sin embargo, no se pueden desechar. Una interpretación interesante es que, aunque algunas SMN repuestas son necesarias para la corrección de la atrofia muscular espinal, no son suficientes, y son requeridos mayores niveles de SMN en el tejido o en los tejidos que no sean del sistema nervioso central (por ejemplo, del sistema nervioso autónomo, muscular, del corazón e hígado) con tal que la terapia en la atrofia muscular espinal pueda ser eficaz. Los experimentos están en marcha para resolver tal problema.
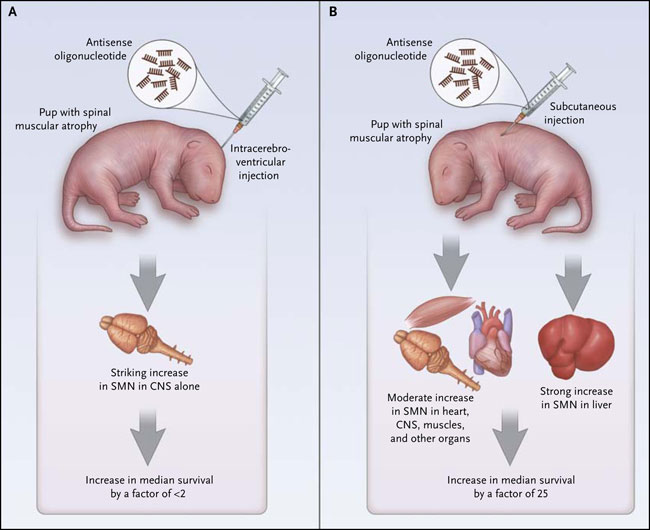
La administración intracerebroventricular de ASO en el sistema nervioso central (SNC) el día 1 tiene un efecto modesto en la supervivencia de animales con atrofia muscular espinal, a diferencia del efecto más profundo con una dosis más grande, y dividida de forma sistémica durante los días 1 y 3 por inyección subcutánea o intraperitoneal.
Por lo tanto, ahora se une el ASO que puede superar a la terapia génica como enfoque primordial experimental en el tratamiento de la atrofia muscular espinal. Una barrera importante son las altas concentraciones de ASO sugeridas. Y para ser más eficaz, los ASO probablemente tendrán que darse en el período post-natal (y pre-sintomático), lo que haría necesario el diagnóstico de la atrofia muscular espinal en recién nacidos. Los avances en tecnología genética que han permitido el cribado neonatal también se pueden utilizar en pruebas pre-natales, que en última instancia, podrían disminuir el número de niños afectados.
El concepto de rescate parálogo - es decir, el tratamiento de una enfermedad genética mediante la modulación de un gen que funcionalmente compensa (al menos en parte) al mutado, no es nuevo pero no está debidamente evaluado. El único ejemplo en el uso clínico es la inducción farmacológica de la hemoglobina fetal para las hemoglobinopatías. Una segunda instancia está en desarrollo: la inducción de la utrofina para tratar la distrofia muscular de Duchenne.
Del mismo modo, si bien la terapia con ASO se ha propuesto para trastornos infecciosos, oncológicos y genéticos, durante casi dos décadas, la única actualmente en uso (en retinitis por citomegalovirus) fue aprobada por la Food and Drug Administration hace 14 años. La siguiente aplicación terapéutica probablemente se apruebe en la apolipoproteína B para la reducción de los niveles de colesterol. Una parte importante de mutaciones raras causantes de enfermedades afecta al empalme, y muchas de ellas parecen manejables con la modulación de ASO. La modulación de SMN2 vía ASO para la atrofia muscular espinal está siendo estudiada en un ensayo clínico, lo que permitiría una transición exitosa a la clínica y por lo tanto mantener la esperanza en personas con atrofia muscular espinal, representando una importante aplicación de concepto en el rescate parálogo y en la terapia con ASO, así como en el estudio de casos en muchos otros trastornos hereditarios que implican mutaciones en los sitios de empalme.
Fuente bibliográfica
Sense in Antisense Therapy for Spinal Muscular Atrophy
Alex MacKenzie, M.D., Ph.D.
Children’s Hospital of Eastern Ontario and the University of Ottawa, Ottawa.
N Engl J Med 2012; 366:761-763