El componente genético de la diabetes tipo 2
El noventa por ciento de los diabéticos padecen diabetes tipo 2. En esta condición, la resistencia a la insulina genera inicialmente un aumento de la producción de insulina por las células beta del páncreas, con tal de prevenir la hiperglucemia. Gradualmente, sin embargo, las células beta comienzan a fallar. La reducción resultante en la producción de insulina, junto con la continua resistencia a la insulina, conduce a la hiperglucemia.
La evidencia de numerosos estudios epidemiológicos demuestra que los factores que predisponen a la obesidad, a la diabetes tipo 2 y a la resistencia a la insulina, son de carácter genético, así como de origen medio ambiental. Las estimaciones para las bases genéticas de la variación fenotípica en la obesidad fluctúan de un 40% hasta el 70%. Los estudios familiares de segregación también muestran que el riesgo de por vida de desarrollar diabetes tipo 2 es del 40% en los descendientes de uno de los padres y de un 70% si ambos padres tienen la enfermedad.
Células beta, estrés y diabetes tipo 2
La causa de la diabetes mellitus tipo 2 corresponde a una combinación de factores genéticos y ambientales que se traducen en la función anómala de la insulina en sus sitios de acción y una disminución de la capacidad de las células beta del páncreas para elevar la secreción hormonal en respuesta al aumento de los niveles de glucosa en la sangre. La variabilidad genética que causa la susceptibilidad a la diabetes era prácticamente desconocida hasta el advenimiento de los estudios de asociación genómica. En 2007, uno de estos análisis identificó relaciones entre la diabetes tipo 2 y seis loci cromosómicos diferentes. Posteriormente, se confirmaron estas asociaciones, pero además también y se identificó una nueva asociación con la variante CDKAL. Estudios posteriores permitieron aumentar a 40 el número de loci implicados. Por desgracia, esta abundancia de lugares no ha mejorado nuestra comprensión de los mecanismos patológicos, en parte porque los estudios de asociación genómica a menudo implican a genes (o regiones no génicas) de función desconocida, como CDKAL1.
En ocasiones, los descubrimientos en diferentes campos se unen para ofrecer nuevos marcos conceptuales que han podido mejorar nuestra comprensión de la diabetes. Un reciente ejemplo lo constituyen dos estudios realizados por Simon Arragain y colaboradores (J Biol Chem 2010; 285:28425-28433) y Fan-Yan Wei y colegas (J Clin Invest 2011; 121:3598-3608), que ligaron a CDKAL1 con la traducción de proteínas y demostraron cómo esta relación puede ser clave para la diabetes tipo 2.
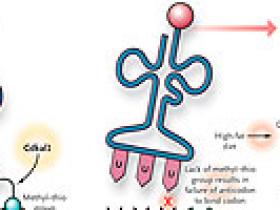
Se había establecido previamente que algunos ARN de transferencia de bacterias y eucariotas presentaban una fracción específica (el grupo metil-tio ms2t6) unido al residuo de adenosina que se encuentra junto al anticodón (figura 1). La presencia de esta fracción es esencial para la fiel traducción de ARN mensajero a proteína. El grupo de S. Arragain informó que CDKAL1 correspondía a una enzima (un metiltiotransferasa) y que se añadía una mitad a los residuos de adenosina. En un ensayo más reciente, el grupo de Fan-Yan Wei concluyó que CDKAL1 se une a un ARN de transferencia específico (tRNALys (UUU) [Figura 1]) añadiendo un residuo de lisina durante la síntesis proteica (es decir, durante la traducción del ARN mensajero a proteína) (figura 2). En otras palabras, CDKAL1 parece tener un efecto global en la producción de proteínas al garantizar la fiel traducción del codón AAA (que codifica lisina) a través de la mediación de un evento altamente específico (la modificación de un residuo de aminoácido específico de tRNALys [UUU]). Los investigadores silenciaron a Cdkal1 en células beta de ratón y observaron una menor respuesta a la insulina cuando se inyectaba glucosa de forma intraperitoneal. Estos hechos se agravaron en ratones alimentados con una dieta alta en grasas.
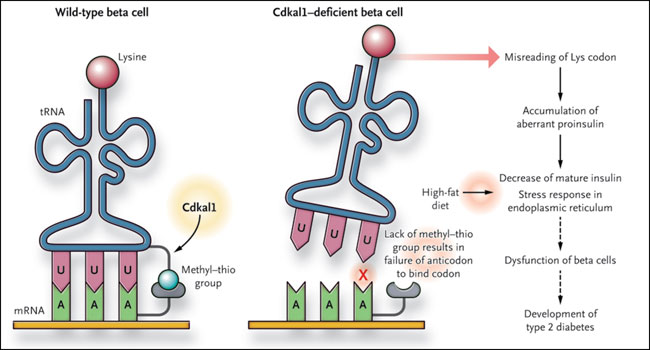
La diabetes tipo 2 está asociada con la variante CDKAL1. Recientemente se le atribuye la siguiente función a este gen: es capaz de modificar un ARN de transferencia específico (ARNt) al catalizar la adición de una fracción de metil-tio (ms2t6) a un residuo adyacente al anticodón (en rosa). Los ratones deficientes en Cdkal1 en células beta incorporan menos residuos de lisina en la proinsulina, lo que contribuye al desarrollo de diabetes tipo 2.
¿Cómo la inhibición de Cdkal1 en células beta genera una débil respuesta a la insulina? Una de las hipótesis más atractivas es la aparente incapacidad de las células beta mutantes para procesar la proteína proinsulina en insulina. Los autores encontraron que la proinsulina en mutantes tenía menor contenido de lisina que la proinsulina de células normales. También determinaron que los niveles del péptido C (un subproducto del procesamiento de la proinsulina) fueron menores en los islotes y en el suero de los ratones mutantes. La vinculación de estas dos observaciones nace del hecho que la lisina constituye el punto de corte entre el péptido C y la cadena A de la insulina (figura 2). Por tanto, una deficiencia de lisina en la proinsulina podría predecir el resultado de una molécula que es resistente a la ruptura en la unión entre el péptido-C y la cadena A.
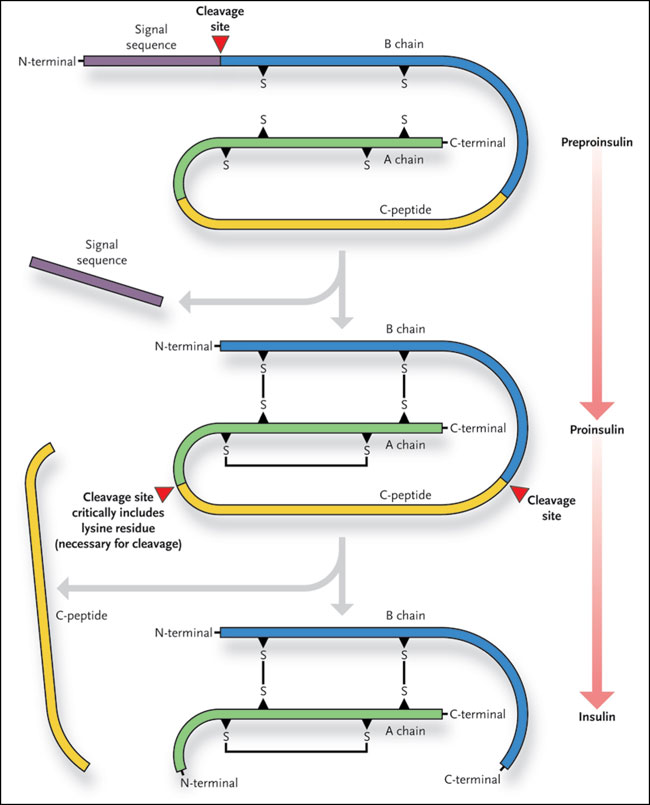
La producción de insulina madura se lleva a cabo dentro de la célula beta y depende de la división de las moléculas de preproinsulina y proinsulina. El sitio de corte en la unión de la cadena A y el péptido C contiene un residuo de lisina, que es fundamental para la ruptura.
Sin embargo, está claro que la salud general de la célula beta mutante también se ve comprometida. Por ejemplo, los autores observaron un aumento en la expresión de moléculas de estrés en el retículo endoplásmico. Este estrés probablemente lo cause un número creciente de proteínas (incluyendo insulina) que son mal plegadas, ya que son deficientes en lisina. Por otra parte, existe escasa superficie celular del receptor de GLUT2 en la célula beta mutante (GLUT2 transporta la glucosa extracelular en la célula, lo que pone en marcha una cadena de acontecimientos que culmina en la movilización de los gránulos que contienen la insulina). Todos, algunos o ninguno de estos eventos pueden ser cruciales para el fenotipo de la diabetes en los ratones mutantes.
Los autores insisten en la necesidad de comprender cómo la variante CDKAL1 puede causar susceptibilidad a la diabetes tipo 2. Varias de las preguntas podrían ser resueltas en futuros estudios. Los investigadores proponen que la falla de incorporar lisina genera el mal plegamiento de la proinsulina e impide el procesamiento proteolítico. ¿Si este es el caso, qué aminoácidos pueden sustituir a la lisina? Si una deficiencia general de la incorporación de lisina en la proteína provoca mal plegamiento de proteínas, tal vez la respuesta celular afecte a la producción de insulina. En la actualidad, hay una serie de ejemplos en los que el plegamiento deficiente en la célula beta evita el procesamiento y el tráfico adecuado de proinsulina entre el retículo endoplasmático y el aparato de Golgi, provocando estrés y una alteración en la estructura mitocondrial. Ya que CDKAL1 se expresa fuertemente en el retículo endoplásmico, su propio mal plegamiento (suponiendo que se produzca) puede afectar a otros plegamientos, procesamientos o al control de calidad, pudiendo causar una acumulación de proteínas y la muerte de las células beta. En un aspecto más amplio, existen muchas asociaciones entre el estrés oxidativo y el fracaso de las células beta. Es interesante considerar que CDKAL1, debido a su composición, pueden ser muy sensible a la oxidación. Como ahora se conocen los mecanismos moleculares de CDKAL1, las investigaciones podrían considerar pequeñas moléculas mediadoras que eviten la progresión de la diabetes tipo 2 en pacientes que llevan variantes de riesgo de CDKAL1.
Fuente bibliográfica
Beta-Cell Failure, Stress, and Type 2 Diabetes
Randal J. Kaufman, Ph.D.
Center for Neuroscience, Aging, and Stem Cell Research, Sanford-Burnham Medical Research Institute, La Jolla, CA.
N Engl J Med 2011; 365:1931-1933