Terapia de reemplazo enzimático para la enfermedad lipídica
Los lisosomas son vesículas situadas en el interior de la célula que poseen diferentes enzimas hidrolíticas, cuya función es degradar proteínas, polisacáridos y lípidos. Las mutaciones de genes que codifican estas enzimas son causa de enfermedades de almacenamiento lisosomal que conducen a la acumulación de sustratos, produciendo efectos patológicos sobre el metabolismo. Particularmente, mutaciones en LIPA, gen codificante para la lipasa ácida lisosomal, conllevan a la acumulación de lípidos en estas estructuras intracelulares, debido a la deficiencia enzimática, perjudicando seriamente el metabolismo lipídico. Si bien, las opciones terapéuticas para esta deficiencia son actualmente limitadas, nuevas investigaciones utilizando proteínas recombinantes han mostrado ser prometedoras y han logrado revertir la acumulación de lípidos en hepatocitos y frenar el daño hepático.
Terapia para la deficiencia de lipasa ácida lisosomal
Existen más de 50 enfermedades diferentes de almacenamiento lisosomal, trastornos genéticos caracterizados por la acumulación de sustratos. Los fenotipos de la patología varían ampliamente, dependiendo de los tipos celulares específicos que sean afectados. La terapia de reemplazo enzimático, ha sido innovadora para aliviar algunas de estas alteraciones. Sin embargo, solamente está aprobada y comercialmente disponible para 7 tipos de complicaciones lisosomales.
El principio de la terapia de reemplazo enzimático es que después de la administración de una enzima recombinante, esta es captada por las células diana y dirigida hacia a los lisosomas, donde puede hidrolizar su sustrato. En la enfermedad de Gaucher, la enzima es asimilada por macrófagos a través de sus receptores de manosa. Por otro lado, en la enfermedad de Fabry, esto ocurre mediante los receptores de manosa 6-fosfato en podocitos y cardiomiocitos, mientras que lo mismo ocurre para la enfermedad de Pompe, pero en cardiomiocitos esqueléticos. En estos trastornos, la cantidad de enzima disponible en los tejidos blanco, a menudo se reduce debido a la absorción inespecífica por los hepatocitos del hígado. No obstante, no existe actualmente una terapia dirigida específica a los hepatocitos.
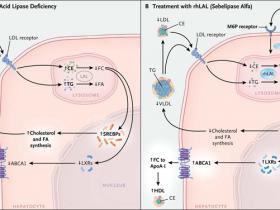
La deficiencia de lipasa ácida lisosomal es una enfermedad de almacenamiento autosómica recesiva causada por mutaciones en LIPA, gen codificante para la enzima lipasa ácida lisosomal, ejerciendo importantes efectos patológicos sobre el metabolismo lipídico en hepatocitos (fig. 1A). La falla en la hidrólisis eficiente de triglicéridos de ésteres de colesterol y triglicéridos en lisosomas hepáticos conduce a la acumulación de lípidos neutros en estas estructuras celulares (fenómeno conocido históricamente como enfermedad de almacenamiento de éster de colesterol), además de provocar hepatomegalia, disfunción hepática, fibrosis y progresión a cirrosis. La dislipidemia también es vista muy comúnmente en pacientes con sustanciales aumentos en los niveles de lipoproteína de baja densidad (LDL) y un grado reducido de lipoporteína de alta densidad (HDL) (fig. 1A). Aunque el gen LIPA es abundantemente expresado en el cerebro, ningún fenotipo neurológico se asocia con esta enfermedad. De esta forma, los hepatocitos son el principal tipo celular para intervenciones terapéuticas.
Las opciones para el tratamiento clínico de personas con la deficiencia son actualmente limitadas. Las estatinas no logran reducir la progresión de la enfermedad hepática y en realidad podrían acelerarla mediante el aumento de captación de lipoproteínas a través de los receptores de colesterol-LDL (podría argumentarse algo similar respecto a los inhibidores de la proproteína convertasa subtilisina-Kexin tipo 9 [PCSK9]). Por otra parte, los inhibidores de la absorción de colesterol poseen cierta eficacia sobre los niveles lipídicos y de alanina aminotransferasa hepáticos, en modelos animales (ratón) de la enfermedad, pero la experiencia clínica con estos inhibidores en personas con deficiencia de lipasa ácida es limitada. El trasplante de hígado ha sido utilizado en individuos que tienen progresión a etapas finales de la enfermedad hepática, por lo tanto, el tratamiento con la enzima de reemplazo dirigida a los hepatocitos es un enfoque lógico (fig. 1B).
La lipasa ácida lisosomal recombinante humana, administrada a ratones LIPA-knockout en estadios patológicos avanzados, revirtió la acumulación de lípidos en los hepatocitos y la lesión hepática, además de disminuir los niveles de lípidos en el plasma. Estas observaciones condujeron al desarrollo de una nueva enzima recombinante (sebelipasa alfa), como un potencial tratamiento clínico. Un estudio de prueba de concepto que involucró adultos con la deficiencia enzimática, mostró una reducción de los niveles de transaminasas y de dislipidemia, lo que persistió durante 1 año durante el tratamiento continuado.
Otros resultados provenientes de un ensayo fase 3 aleatorizado y controlado, han probado esta enzima recombinante en 66 adultos y niños con deficiencia de lipasa, muchos de ellos con evidencia de fibrosis hepática avanzada. Los niveles de aminotransferasa plasmática, el contenido de grasa hepático y el volumen del hígado se redujeron significativamente debido al tratamiento. Aunque estos hallazgos sugieren que esta terapia es probable que reduzca la progresión a la etapa final de la fibrosis hepática, se requerirán de seguimientos a más largo plazo en una cohorte más amplia para confirmarlo. Además, los niveles plasmáticos de lípidos mejoraron sustancialmente, con reducciones significativas en niveles de colesterol LDL y un incremento en colesterol HDL (fig. 1B).
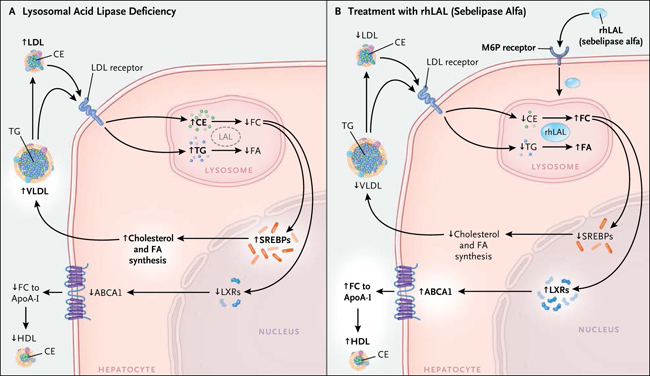
Figura 1: efectos de la deficiencia de lipasa ácida lisosomal hepática sobre metabolismo lipídico hepático y plasmático y Efectos de la infusión de Sebelipasa Alfa
Las lipoproteínas de baja densidad (LDL) y lipoproteína s de muy baja densidad (VLDL), son captadas por los hepatocitos por medio del receptor de colesterol LDL (y otros receptores) y son dirigidos a los lisosomas (Panel A). La ausencia de lipasa ácida lisosomal resulta en una disminución de la hidrólisis y acumulación lisosomal de ésteres de colesterol (CE) y triglicéridos (TG), además de una reducción de la generación de productos que incluyen colesterol libre (FC) y ácidos grasos (FA). El descenso del colesterol libre proveniente de los lisosomas se traduce en un incremento del procesamiento y actividad de las proteínas de unión a elementos regulatorios de esterol (SREBP) y en una acentuada síntesis de colesterol y ácidos grasos, lo que acrecienta la secreción de colesterol VLDL y los niveles de LDL. Adicionalmente, la escasa generación de lisosomas libres de colesterol genera una baja activación de los receptores X del hígado (LXR), una disminución de la expresión del cassette del transportador de colesterol de unión a ATP (ABCA1). Por otro lado, esto también reduce el flujo de colesterol a la apolipoproteína AI (ApoA-I), dando lugar a niveles plasmáticos reducidos de colesterol HDL. Luego de la infusión intravenosa de lipasa ácida lisosomal humana recombinante (rhLAL) esta es captada por los hepatocitos, por medio del receptor de manosa-6-fosfato (M6P) y dirigida hacia los lisosomas (Panel B). La enzima recombinante compensa la deficiencia e hidroliza los ésteres de colesterol y triglicéridos, lo que resulta en la reversión de los defectos metabólicos de lípidos intracelulares y del plasma.
Existe evidencia anecdótica de que el riesgo de enfermedad cardiovascular aterosclerótica puede aumentar entre pacientes con deficiencia de lipasa, al menos en parte, debido a la dislipidemia. Estudios en ratones han demostrado que las infusiones de enzima recombinante humana es capaz de revertir la aterosclerosis. En consecuencia, será interesante determinar si el tratamiento con sebelipasa alfa no sólo mejora los perfiles de lípidos sino que también si tiene la capacidad de reducir el peligro cardiovascular entre quienes padecen esta alteración metabólica.
La deficiencia enzimática es difícilmente reconocida y puede ser mal diagnosticada como hipercolesterolemia familiar o hígado graso no alcohólico. Debido al efectivo reemplazo, esta terapia puede estar prontamente disponible, por lo que los médicos necesitarán, tan pronto como sea posible, examinar y diagnosticar este desorden e iniciar su tratamiento. La deficiencia enzimática debe sospecharse en casos de hipercolesterolemia sustancial sin historial familiar sobre todo si es acompañada por un bajo nivel de colesterol HDL, elevado nivel de aminotransferasa, o hígado graso. También es de relevancia cualquier paciente con un diagnóstico de cirrosis micronodular en una biopsia hepática. Para esto, un simple ensayo enzimático sanguíneo se encuentra clínicamente disponible para individuos en los que se presume el diagnóstico.
Los individuos con las mutaciones inactivantes más marcadas del gen LIPA tienen severas dificultades para mejorar su pronóstico en la infancia temprana y poseen una progresión mortal hacia el primer año de vida (una condición históricamente conocida como enfermedad de Wolman). Actualmente, existe un ensayo en curso sobre la lipasa recombinante en cuestión, realizado en este tipo de pacientes (ClinicalTrials.gov, NCT02193867) y los informes iniciales sugieren una respuesta alentadora. La terapia enzimática de reemplazo podría salvar la vida de quienes sufren de esta temprana condición, pero requeriría un pronto diagnóstico e inicio de la terapia, lo que sería ampliamente facilitado por un screening neonatal.
En resumen, esta enzima de reemplazo es la primera terapia dirigida específicamente a hepatocitos. Si es aprobada, puede ser transformar el enfoque en el tratamiento de la deficiencia de comienzo infantil y tardío de la lipasa ácida lisosomal y dar lugar a una mayor conciencia y mejores diagnósticos de este subestimado trastorno genético lipídico.
Fuente bibliográfica
Lysosomal Acid Lipase Deficiency — A New Therapy for a Genetic Lipid Disease
Daniel J. Rader, M.D.
University of Pennsylvania, Philadelphia
DOI: 10.1056/NEJMe1509055