Hemoglobinopatías
Poner un dedo en el interruptor
El estudio de la biología de las células sanguíneas rojas ha proporcionado un terreno fértil para la realización de ensayos celulares clásicos, bioquímicos, genéticos y moleculares. Descifrar cómo los cambios de desarrollo en la expresión de la cadena de hemoglobina son controlados ha sido de particular interés, debido no sólo por la curiosidad acerca de los mecanismos básicos, sino también por el deseo de aliviar la morbilidad asociada a las hemoglobinopatías, el trastorno más común de un único gen en todo el mundo.
Se sabe que la transición de la expresión de la β-globina fetal a β-globina adulta es un paso clave en la maduración del linaje de los glóbulos rojos. Nuevos estudios muestran que la proteína de dedos de zinc KLF1 (también llamado EKLF) utiliza medios directos e indirectos para moderar un “interruptor” que regula y controla finalmente la expresión de la globina fetal hacia la forma adulta y que la pérdida mono alélica de la expresión de KLF1 conduce a la persistencia de la hemoglobina fetal. Los estudios también han descubierto una relación normativa entre KLF1 y un segundo factor de transcripción, BCL11A, que ayuda a explicar cómo KLF1 organizar este interruptor.
Alterando los cambios de la hemoglobina
Las observaciones naturales proporcionan pistas importantes sobre cómo las terapias pueden desarrollarse para mejorar una enfermedad. Trastornos de la β-hemoglobina, incluyendo la enfermedad de células falciformes y la β-talasemia, son buenos ejemplos de esta máxima, ya que una gran variedad de observaciones clínicas han demostrado que niveles elevados de hemoglobina fetal son capaces de aminorar la gravedad de estas enfermedades. Tal observación ha llevado durante décadas a la caza de estrategias que aumenten la expresión de la hemoglobina fetal de forma selectiva. Existe cierto éxito en el desarrollo de fármacos para lograr dicho incremento para beneficio terapéutico, como lo demuestra el uso de la hidroxiurea. Sin embargo, las bases moleculares de la eficacia de estos agentes siguen siendo desconocidas, y han demostrado ser moderadamente eficaces en los subgrupos de pacientes analizados.
A lo largo de la gestación humana, la hemoglobina que se expresa en el feto es predominante hemoglobina fetal, que se compone de dos cadenas de α-globina y dos cadenas de γ-globina. Durante el nacimiento, hay un cambio de hemoglobina fetal a hemoglobina adulta, mediado por una permuta de γ-globina a β-globina adulta producida por el locus del gen β-globina (figura 1). En la infancia, a mediados y finales de la vida, la producción de γ-globina constituye sólo una pequeña fracción de la producción de β-globina adulta. Como resultado, la hemoglobina fetal corresponde a menos del 1% del total de hemoglobina. Aunque los mecanismos moleculares que median dicho cambio en la ontogenia humana han sido ampliamente investigados, hasta hace poco las bases moleculares de esta transformación han permanecido desconocidas.
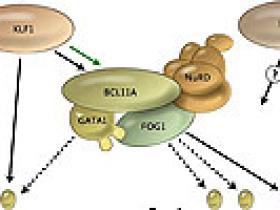
Un reciente estudio realizado por Dewang Zhou y colegas (Nat Genet 2010; 42:742-744) proporciona una nueva visión de este tipo de mecanismos mediadores de cambio en el desarrollo, al demostrar que el factor de transcripción KLF1 (factor de Krüppel tipo 1) es un regulador importante de tal proceso. Estudios de asociación genómica han generado más ensayos para identificar los mediadores moleculares de este mecanismo. El lugar implicado se encuentra dentro del gen que codifica el factor de transcripción BCL11A (linfoma de células B/leucemia 11A), que ha dado lugar a trabajos que muestran que BCL11A es un importante regulador del desarrollo del interruptor de la hemoglobina fetal a adulto en el ser humano, sirviendo como un represor del gen de la γ-globina. BCL11A parece actuar directamente sobre el locus de la β-globina, junto con un conjunto de proteínas asociadas. Sin embargo, se desconocen los factores que permiten la regulación del desarrollo de la expresión BCL11A y que puedan también afectar otros aspectos de la regulación génica de la globina.
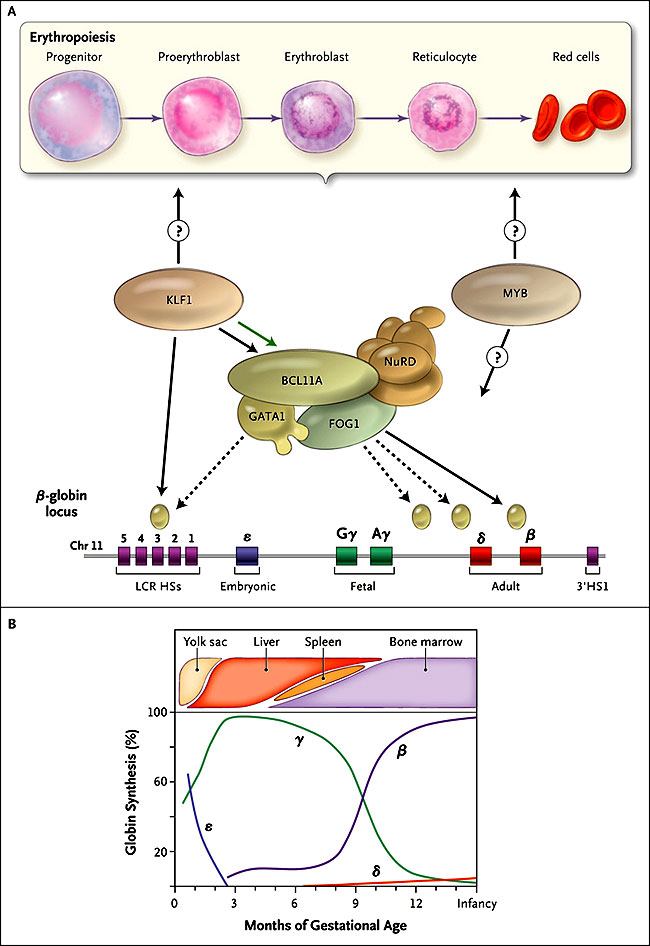
El locus humano del gen β-globina, que se encuentra en el cromosoma 11, tiene una serie de cinco genes que son expresados en diferentes fases de la ontogenia humana (parte inferior del panel A). El gen ε-globina se limita al control de los glóbulos rojos producidos en las primeras semanas de gestación. A su vez, los genes de γ-globina codifican los genes que únicamente forman la hemoglobina fetal, que es la que predomina durante el curso de la gestación. Hay un “interruptor” para la expresión del gen β-globina adulta durante el momento del nacimiento (panel B). El linfoma de células B/leucemia 11A (BCL11A) y sus proteínas socias, incluyendo la proteína de unión GATA 1 (GATA1), proteína de dedo del cinc multi-tipo 1 (ZFPM1 o FOG1), y la remodelación de nucleosomas y complejos deacetilasa (NuRD), se unen a secuencias dentro del locus de la globina (se muestran como óvalos en el panel A) y reprimen la expresión de los genes de γ-globina. El factor de Krüppel tipo 1 (KLF1) interactúa con este proceso mediante regulación positiva para la expresión de BCL11A (flecha verde en el panel A) y también por unión directa y promoviendo la transcripción del gen β-globina adulta. Se desconoce si KLF1 afecta la expresión de la hemoglobina fetal a través de más efectos globales sobre la eritropoyesis (parte superior del panel A), lo que también pudiese ser objetivo de otros factores asociados con la hemoglobina fetal (por ejemplo, MYB).
El grupo de D. Zhou comenzó a desarrollar un modelo de ratón con bajos niveles de expresión KLF1, para evitar la mortalidad temprana ya que se había visto en modelos de ratón que tenían una anulación completa de este gen. La inhibición de la expresión de KLF1 en ratones, permitió a los investigadores observar un marcado aumento en la expresión de los genes de globina de embriones de ratón (que están evolutivamente relacionados con los genes de la hemoglobina fetal) y los genes de γ-globina humana (de un locus transgénico de β-globina humana). La expresión de BCL11A en estos ratones estuvo notablemente reprimida, en comparación con la expresión proteica en los controles de tipo salvaje. Esta observación, junto con la constatación que KLF1 se une directamente al promotor BCL11A, sugiere que KLF1 regula positivamente la expresión de BCL11A. Por otra parte, KLF1 también se une directamente en el lugar β-globina, lo que sugiere que puede regular la expresión de los genes de globina. D. Zhou y colegas también probaron el efecto de la caída de KLF1 en células progenitoras eritroides, observando una reducción en la expresión de BCL11A y un aumento concomitante en la expresión de γ-globina, un hallazgo alentador, dado que los aspectos de la regulación de genes de globina son divergentes entre los ratones y los seres humanos.
También es reconfortante la observación de J. Borg y colegas (Nat Genet 2010; 42:801-5) que una mutación sin sentido en KLF1 se asocia con la persistencia hereditaria de hemoglobina fetal en una familia maltesa, proponiendo que la haploinsuficiencia de KLF1 aumentaría la producción de hemoglobina fetal en vivo sin una eritropoyesis ostensiblemente molesta. Sin embargo, los portadores de esta mutación presentaban niveles muy variables de hemoglobina fetal, que iban desde 3 a 20%, es decir, el papel de KLF1 en estos cambios puede complicarse por la presencia de otros factores que probablemente influyen en el fenotipo.
La reducción de los niveles de KLF1 puede ser una forma importante de aumentar la hemoglobina fetal en los pacientes. Es posible que las moléculas pequeñas o pequeños ARN de interferencia para KLF1 puedan ser eficaces en la inducción de la producción de hemoglobina fetal en los humanos. KLF1 es un candidato particularmente atractivo, debido a la expresión exclusiva en el linaje eritroide. Sin embargo, la precaución es necesaria. KLF1 tiene un amplio papel en la eritropoyesis, y es posible que la interrupción de su función pueda interferir con el proceso. Con esta advertencia en mente, es evidente que es necesario seguir trabajando para determinar la mejor manera de alterar el eje de regulación relativo a KLF1, BCL11A, y la hemoglobina fetal. Los análisis adicionales de esta vía y otros factores, tales como el factor de transcripción MYB, que han sido implicados en el silenciamiento de la hemoglobina fetal en los estudios genéticos pueden alimentar las estrategias focalizadas para la inducción de la hemoglobina fetal.
Fuente bibliográfica
Reversing the Hemoglobin Switch
Vijay G. Sankaran, M.D., Ph.D., and David G. Nathan, M.D.
Children’s Hospital Boston, Dana–Farber Cancer Institute, and Harvard Medical School
N Engl J Med 2010; 363:2258-2260